@yangfch3
2016-06-12T17:19:18.000000Z
字数 9242
阅读 5181
药物化学
化学
---
Author: yangfch3
Date: 06/13/2016
---
绪论
药物研究的历史
- 开始,新药完全来源于经验主义
- 从植物中分离活性成分
- 系统地探索新的合成物,并且以动物作为实验体进行药物生化影响的研究
- 使用分子和其他体外测试系统精确模型替代动物实验
- 蛋白质晶体、分子模型、SAR(特定吸收率)等试验,计算机的支持,新的作用靶的发现,在基因层面考查药物的价值,转录和翻译分析,使用 siRNA 进行基因沉默
- 中国最早的药物
- 神农发现的麻黄(主要活性成分:麻黄素,1897年正式分离),现在,麻黄素的异构体:假麻黄碱用于解充血,也作为非处方感冒药,脱氧麻黄碱现在则是一种兴奋剂。
- 印度药物简史
- 原产自南亚的姜黄(主要作用成分:质量分数为2-5%的姜黄素),从姜黄类植物的根茎处分离得到,被广泛用于咳嗽、肝病、风湿病、炎症和厌食症,并被广泛研究,现在被研究用于 抵抗癌症、抗凝血剂、降胆固醇、抗糖尿病和刺激治疗,现在许多人将麻黄素加入了他们长期的健康饮食计划中用于抵抗各种癌症
- 中东药物简史
- 诺亚的葡萄园和酒(乙醇)
- 古希腊、罗马、拜占庭帝国时期
- 希波克拉底 -- 医学之父,体液药,四元素:空气、土地、火和水;最先的脱离神话与迷信的药物是:吗啡,鸦片的有效成分,从罂粟壳中干燥、提取。吗啡结构改变后,变为了 海洛因 和 可卡因。
- 药物发展史幸运的早期
- 偶然的发现物质的活性,染料与药物在化学工业时代之前就已经大放异彩;弗莱明 发现了第一种 抗生素:青霉素 -- 是真菌作为防卫细菌的产物。
麦角灵生物碱的研究导致了人们发现了 麦角酸二乙基酰胺(LSD) 的致幻效果,最后导致了 麻醉药 的出现 - 经典药物研究
- 病理生理学、细胞、分子知识有限,所以借助动物做实验;在这个阶段,为 传染病、精神病等疾病的治疗做出了巨大的贡献
- 阿司匹林
- 最先是在 柳树皮 中发现了 水杨苷,菲力克斯·霍夫曼提纯了 乙酰水杨酸,之后 乙酰水杨酸 被 拜耳 注册为 阿司匹林,延长了人类的平均寿命。US FDA 发现 ASA 能降低心脏病发作 20%,癌症患病率 60%。之后发现这对鼠有一定的 致畸率
- 疟疾
- 奎宁 -- 金鸡纳碱,金鸡纳树皮活性成分,有较大的治疗效果,也有较大的副作用,但是知道前几年,一直在使用,特别是用在注射上。
DDT 与 DDE 被研发后,疟疾发病率骤降。
从艾蒿中分离的 二氢青蒿素 的衍生物 青蒿素,中国科学家 屠呦呦 使用乙醚提取,发现青蒿提取物对 疟原虫 的抑制率达到 100% - 药物改变了社会结构
- 改善了生活质量
延长了平均寿命(HIV 蛋白酶、逆转录酶,无细胞毒素的抗肿瘤药物) - 为什么药物化学面临更大的挑战
- 1、抗生素的滥用
2、随着人均寿命的延长,肿瘤、神经方面、身体退化、自身免疫疾病开始流行
3、HIV 以及 SARS 类由病毒引发的新疾病
4、避孕药、伟哥等药物的使用 - 药物和药物化学家
- 药物化学家:试图设计并合成有利于人类的药物或药剂
- 好药与坏药
- 好药应该是
1. 无毒、没有有害的副作用
2. 易于使用
3. 能够完成药物设计的初衷 - 有多少好药?
- 市面上还没有完全符合以上好药标准的药物,只有少数药物接近上面的标准
- 青霉素的问题
- 1、不一定对所有已知细菌有效
2、越来越多的细菌具有了耐药性
3、并不完全安全 - 普遍接受的“坏药”
- 吗啡
巴比妥类药物
乙醇 - 一些药物的美丑
-
咖啡因:所有的茶、咖啡、可可内都含有咖啡因,能改变一个人的情绪
尼古丁:有镇定作用
酒精:成瘾,社会问题,难以管控,生物化学反应并不明确总结:我们所认为的常识未必正确
- 药物与毒物
- 盘尼西林对人无影响,但是对细菌细胞是毒药
不能脱离剂量谈药物、毒物 - 食品与药物
- 食品必然会包含有毒化学物质,萝卜、芥菜、樱桃、李子内都含有致癌物质
- 治疗指数(TI)
-
测量指数用于测量药物在 低剂量下的有益表现 与 高剂量下的有害表现 的指标
治疗指数越高,表示:有益与毒物化剂量之间的安全界限越大,越不容易因为一丁点剂量的影响就变为毒物
大麻的指数是 1000,乙醇的指数是 10,说明大麻更加 安全与可预测注意:治疗指数无法指明长期服用所带来的毒性的可能
结论:剂量决定毒性 - 基本概念
- 药物化学重点在 分子水平 上关注 生物活性化合物 的 发现、发展、鉴别、作用方式。重点关注 药物,也关注药物代谢产物、相关化合物的研究、鉴别和合成
- 药物化学
- 研究药物设计以优化药物动力学和药效学,合成新药物
- 药物动力学
- 研究药物在身体各处 浓度-时间 关系的控制因素
- 药效学
- 研究药物与其目标(靶)之间的分子与细胞作用
- 什么是药物
- 广义上来说,药物是任何用于诊断、治疗或预防疾病的物质,它可以是合成的,半合成的,天然的。药物能增强或减弱现有器官、组织或细胞的功能,但是不能赋予它们新功能
- 药物如何起作用
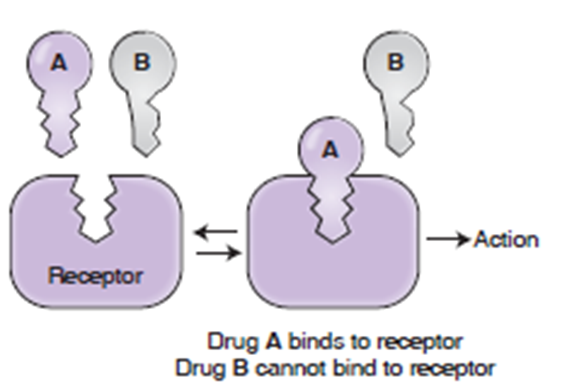
上面是关于 药物-受体 简单的图解。然而实际上,大多数目标(靶)并不是严格地像 钥匙-锁 的关系,活性位点能够改变它的形状和大小- 药物如何设计
- 1、随机筛选(试验-纠错机制,发现先导化合物)
2、合理药物设计(了解疾病的靶标 → 合成结构符合要求的化合物 → 利用计算机建模等手段优化化合物,并且该化合物必须能通过人体的屏障,能够抵抗人体的分解,便于从人体排出) - 如何设计出更好、更安全的药物
- 1、靶标必须是确定的、有效的
2、组织一个虚拟团队
3、研究与应用 ADMET(吸附、分布、新陈代谢、排泄和毒性) 理论
4、确保足够的资金进行前期样品的测试以及临床测试 - 药物使用方法
- 大多数使用口服
少数使用注射
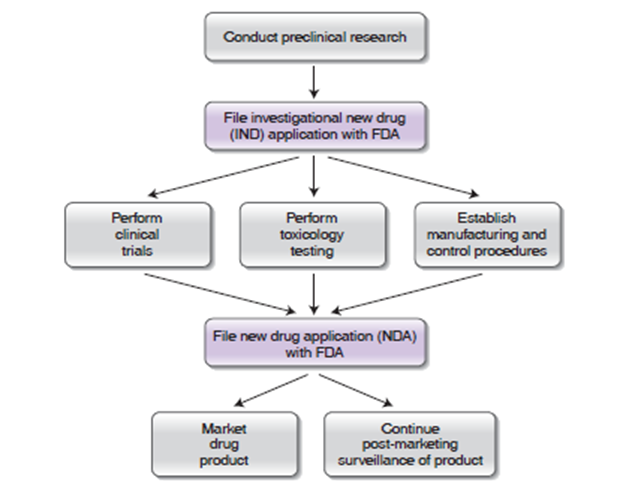
少量采用其他特殊方法 - 药物研发与投入市场的流程
专业词汇
麻黄素 ephedrine
假麻黄碱 pseudoephedrine
脱氧麻黄碱 methamphetamine
姜黄素 curcumin
乙醇 ethanol
吗啡 morphine
青霉素 penicillins
青霉素 aspirin
乙酰水杨酸 acetylsalicylic acid -- ASA
治疗指数 therapeutic index
药物动力学 pharmacokinetics
药效学 pharmacodynamics
药物与靶
- 药物发挥功效必须实现的两个功能
- 到达作用位点
与它们的靶相互作用(一般是人体中的某些物质) - 屏障
- 结构屏障:细胞膜能组织药物从它的进入位置到达作用位点
化学屏障:体液的 PH 和溶解度能影响药物的溶解和离子化
生化屏障与靶:转运蛋白酶,与药物结合的受体,导致药物被摧毁或带来amazing效果 - 影响药物作用的理化性质
- 物理性质:溶解度,分离系数和离子化影响药物的吸收
化学性质:共振结构、诱导效应可能导致药物与靶或其他蛋白质结合
立体化学性质:形状、大小影响药物如何与靶相互作用 - 药物作用靶标
- 酶
受体
转运系统
细胞信号系统
细胞蛋白质合成系统
储存位点 - 互补性
- 1、药物必须能与靶适配以产生靶标响应
2、药物和它的靶标必须有互补的三维结构
3、大部分药物靶标是体内的蛋白质,除了抗生素等药物 - 蛋白质的生物必须性
- 蛋白质是人体细胞、组织、器官的结构、功能和管理的必需品,它们必须能产生合适的蛋白质,更改它们需要蛋白质的组成和数量会导致细胞的死亡
- 药物-蛋白质相互作用
- 许多现在的药物都是与蛋白质的结构适配,蛋白质是药物作用的靶标
- 蛋白质靶标的种类
- 受体
酶
离子通道
转运器 - 药物的药效基团
- 药效基团与靶标必须在物理、立体结构上匹配
- 向量基团(Vector Groups)
- 向量基团:不会与靶标结合,但是将药物导向作用位点,帮助减小毒性
- 携带基团(Carrier Groups)与弱势基团(vulnerable groups)
- 携带基团:控制电离、亲油性,影响药物的吸收、分布和代谢
弱势基团:对酶促作用敏感,决定药物在体内的新陈代谢 - 生物电子等价性
- 化学电子等价性 指的是,由于电子结构相同,导致离子、化合物或元素的物理化学性质相同
生物电子等价 是电子等价在生物系统上的应用,能指导我们对分子进行修饰以获得我们渴望的物理化学性质或生物作用
生物电子修饰 的主要作用
(1)保持原来的生物活性,但是提高和改善药物动力学上的理化性质
(2)保持他们的理化性质,但是增强或改善药物的生化效应
生物电子修饰 手段:引入取代基、桥基、成环原子或集团、大基团
专业词汇
屏障 barriers
受体 receptors
生物电子等价性 bioisosterism
配体和受体
细胞与器官之间的信号系统主要包括:配体 和 受体
- 配体
- 细胞释放的与受体作用的化学物质
- 受体
- 与配体有生物互补性,有与配体结合的作用位点,细胞内或细胞上大分子物质
- 细胞信号
- 细胞释放、转运、接收、响应环境信息,这些信息由内源性配体(又叫做 细胞介质 和 信号分子)携带。人体细胞同样可以侦查外部的药物、细菌、病毒度毒性信号。
- 信号处理流程
- 1、基于接收到的信号,信号细胞合成适当的配体
2、信号细胞释放配体分子到细胞外液
3、停留在外液或配体分子被转运到靶细胞
4、靶细胞 上的对应 受体 识别并结合 配体,组成复合物;受体 可能在靶细胞的内部也可能在细胞膜上
5、配体-受体 复合物将信号转变为细胞内信号,触发靶细胞内的生化反应
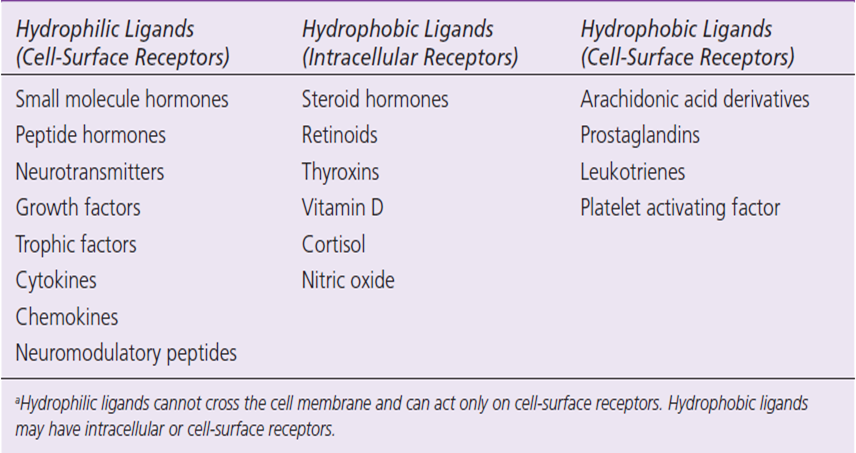
6、配体被移除,结束响应 - 经典的内源性配体
- 受体与配体的相互作用
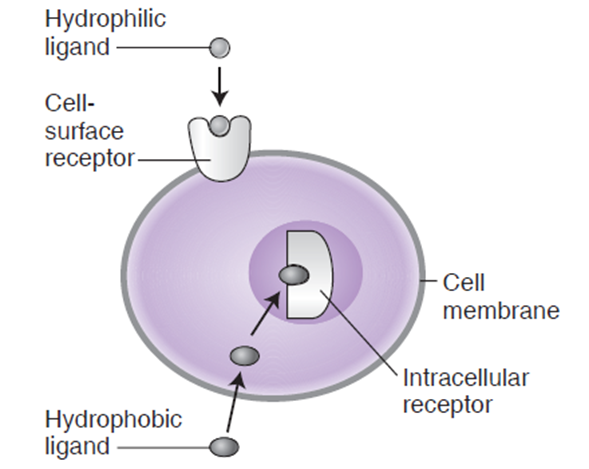
(1)识别:受体侦测到配体,并与之形成复合物
(2)信号转换:配体-受体 结合物导致细胞发生一系列变化,做出生物反应- 非催化型受体
- 配体-受体 结合是可逆的,作用完成后便分离(非催化型受体是药物作用的基础,又叫 非共价键药物)
- 催化型受体
- 一些 非催化剂受体 能在配体缺少的情况下保持持续兴奋,但当配体与之结合后能抑制其持续兴奋,这样的配体属于 抑制剂
配体能与 触媒受体 在细胞内或细胞膜上结合,这时,配体 就被视为 底物,酶与底物形成复合物,并且酶将配体催化为性质不同的产物,然后释放 - 激动剂与拮抗剂
- 配体 与受体作用导致细胞内发生变化,并带来生物反应的叫做 激动剂
配体 与受体形成复合物,但是不产生分子内的附加变化、不造成生物反应的叫做 拮抗剂 - 受体类型集合
- 受体类型集合具有与配体 相似的结合域,但是又 不同的信号转导域,受体类型集合内的受体具有一定的同一性
所以,我们的药物有两类情况:
(1)专一性:专门针对一个受体类型,而不针对受体类型集合,药物选择性高
(2)普适性:一个药物可以结合许多相似的受体类型,药物用途广泛但是会导致不必要的副作用 - 信号转导过程
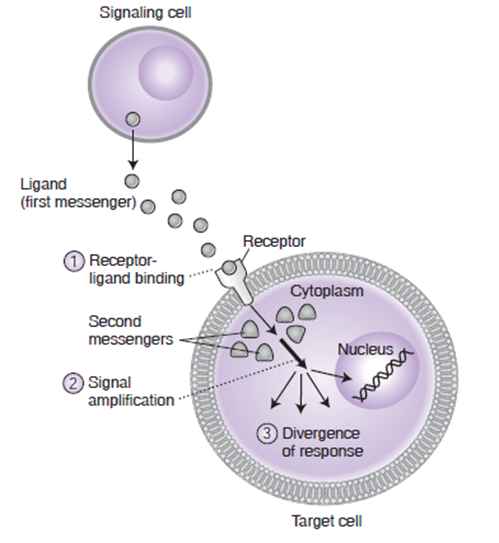
- 受体分类
- 细胞表面受体
1. 离子通道受体
2. G蛋白偶联受体
3. 酶受体
细胞内受体
1. 转录受体
2. 细胞内的酶 - 竞争性拮抗剂与非竞争性拮抗剂
- 都能减小或消除配体-受体结合后带来的生化反应
- 化学拮抗剂
- 化学拮抗剂可以与激动剂反应,改变其结构,从而导致其无法与受体复合,使药物失活;化学拮抗剂可以降低活性位点处激动剂的浓度,从而治疗剂量过多或中毒反应
- 药物作用的立体选择性
- 1、对映异构体1是活性的,对映异构体2也有同样的活性
2、1是有活性的,2是无活性的
3、1是激动剂,2是同样受体的拮抗剂
4、1是有活性的,2有不一样的活性
5、1是有活性的,2是有毒的 - 酶抑制剂
- 能与酶结合降低或者废除催化活性的物质,有可逆的酶抑制剂,也有不可逆的酶抑制剂
药物研发
- 靶标的确立与有效化
- 先导化合物的生产
- 先导化合物的优化
- 早期临床试验(一期,二a期)
- 后期临床试验(二b期,三a期)
靶标的确立和有效化
人类基因组计划
蛋白质计划
仿创结合,根据现有药物的缺陷进行创新
药物化学家不会过多的参与到靶标的发现的过程中
先导化合物的发现
- 什么是先导化合物
- 对于受体具有活性与选择性
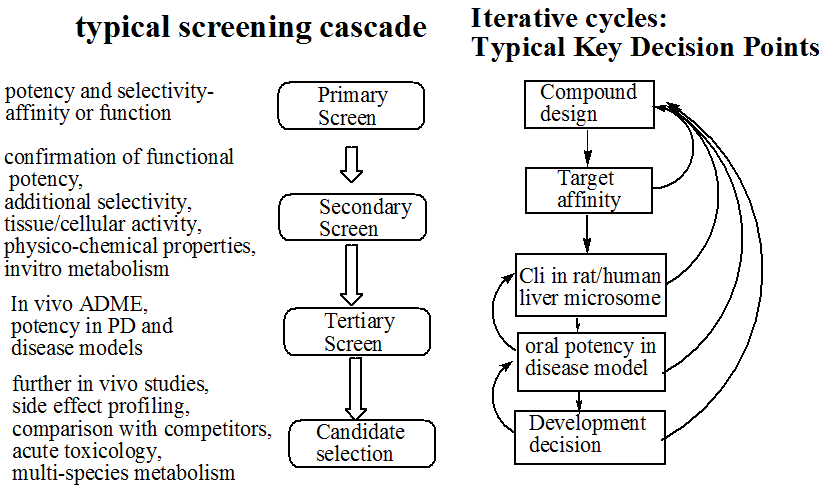
先导化合物的来源
天然产物
- 从天然物质中获取
- 陆地资源:细菌、真菌、植物、昆虫等
- 海洋资源:海藻、珊瑚、海洋植物
- 民俗药物,民间医学(中医)
- 从天然物质中获取
- 已有的药物(me-better) -- 使用 SOSA
- 更有效,生物利用度更高
- 消除副反应(☆)
- 更好的剂型、药前体
- 合理药物设计
- 理解疾病的生理基础/生物过程
- 明确地药物设计出发点
- 生物测定其活性
- 人体试验
- 意外发现
- 合理药物设计
-
分为
基于结构的药物设计(计算机虚拟辅助)
基于性质的药物设计:活性与选择性是最重要的影响因素,但不是决定因素- 活性 ❤
- 选择性 ❤
- 有效性
- 医学需要
- 市场竞争与抢占潜在市场
- 毒性基团
- 一些官能团往往在引入药物后可能造成整体分子的毒性,因而,这些官能团在引入药物分子中时是有所保留的。这些基团叫做毒性基团(toxicophores)
- 活性与选择性
-
活性的要求因药物作用的分子靶的不同而不同,大多数酶抑制剂(inhibitor),GPCR 的拮抗剂(antagonist)或者激动剂(agonist)活性要求达到 nM 级, 而离子通道调节剂则 µM 级就可以了
选择性要求一般在30~50倍,最低要求10倍
先导化合物的优化
- 基于性质的设计优化
- ADME:吸收度、分布系数、新陈代谢、排泄,几个方面的优化
- 两个主要的优化手段
- 1、药理学优化(药效、选择性)
2、ADME/Tox - 化合物的属性会影响以下
- 类药性赋予化合物好的 ADME/Tox 特色
药物化学家通过结构修饰控制性质
生物学家利用性质优化生物实验 - 类药
- 拥有良好的 ADME/Tox 性质,通过了针对人体的临床实验第一阶段的药物
- 先导化合物研究的进步
- 1、药理学筛选从体内转移到体外
2、初始优化已经基于物质结构大数据
3、计算机虚拟化可以用于化合物设计
4、先导化合物的优化从单线程到多线程并行
5、更加微量化 - 可以着手研究的性质
- 结构性质(氢键、亲油性、形状、分子量)
物理化学性质(溶解度、渗透性、化学稳定性、反应)
生化性质(新陈代谢、蛋白质、组织作用)
药物动力学/毒性(生物利用度、半衰期、)
化合物的结构决定药物动力学与毒性,性质的短缺会导致药物效果的大打折扣
结构 - 活性/性质关系方法(Structure Activity-Property Relationship,SAR/SPR)
药物研究的成功是综合各方面均衡的结果
口服方式的药物会遇到一系列稳定性的挑战:分泌系统到内分泌系统之间的屏障
体外实验也会遇到一系列的挑战
- 药物路由途径
- 口服
皮下
局部
肌肉
脑膜
坐药
鼻子
含片
舌下
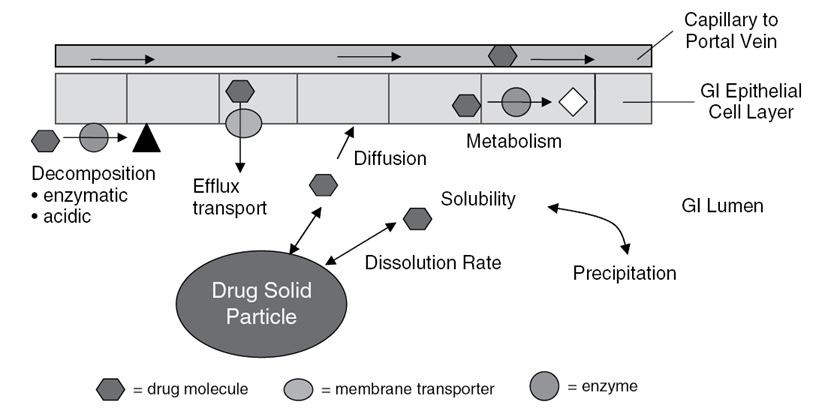
... - 口腔和胃部屏障
- 胃部停留时间段,经由胃部的血流少
胃酸分解,PH 较低(1-2),可能导致药物水解失活
口腔和胃部的酶可能造成药物分解
十二指肠、空肠、回肠 PH 逐渐升高,回肠为 中偏碱
药物能通过肠道壁(两层亲水-疏水层)扩散到内分泌系统中
- 肠细胞内部的新陈代谢
- CYP3A4 - 细胞色素P450酶3A4 是主要的新陈代谢酶,在 CYP3A4 催化下的新陈代谢是药物进入人体循环之前的第一次代谢,细胞内的 Pgb 可以催化分解药物,以降低细胞内的药物浓度。
代谢一阶段:NADPH → NADP+
可以阻塞代谢位点
移除不稳定的功能基团
代谢第二阶段:在 UDPGA 的作用下添加一个大的记性基团
代谢的主要场所:肝脏
药物分子可以从小肠上皮细胞渗透到血液中
- 肠道中的屏障
- 血流中的屏障
- 被血浆中的酶水解
被血浆中的蛋白质结合
被红细胞结合
肝脏中的屏障
肾脏中的屏障
一些器官有屏障,能阻碍药物进入器官中,胎盘、睾丸、脑 都有这样的屏障。
血脑屏障 BBB
- 分子手性对于屏障和性质的影响
- 溶解度
传输体的绑定
代谢
血浆蛋白的绑定
毒性 - 生物屏障总览
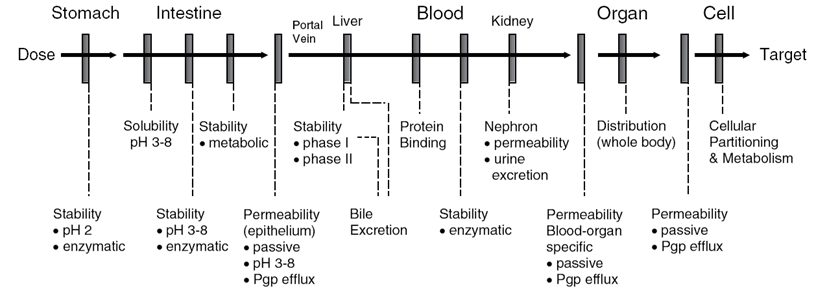
pH
酶催化
代谢- 剔除 类药分子的五条基本法则(便记、易用、正确、广泛应用、基于大量的研究、很好的工作)
- 1、 多余五个氢键给体
2、分子量大于 500
3、脂水分配系数的对数值(logP)logP > 5
4、多余十个氢键受体
5、生物转运蛋白不需要满足以上规则
(以上又叫:Lipinski “5” 原则) - 附加的 Veber 规则:更适于口服的条件
- 化合物中可旋转的键的数目不超过 10 个
总共不多于 12 个氢键(给体+受体) - Oprea “3” 原则
- 1、总分子量 ≤ 300
2、logP ≤ 3
3、可旋转的键 ≤ 3
4、氢键给体 ≤ 3
5、氢键受体 ≤ 3
6、分子表面积 ≤ 60 cm^2 - logP 和 logD
- 脂水分配系数以P表示,化合物在有机相中浓度Co,在水相的浓度Cw;P=Co/Cw
logD 是在不同 pH 下的 logP - 影响 logP 的结构因素
- 分子体积
偶极性
氢键酸性
氢键碱性 - 亲油性改变的条件
- 溶剂
pH
离子强度
缓冲溶液
共溶溶质或共溶溶剂影响
logP 处于 0~3 时亲油性与亲水性都达到最优
处于 1~3 时,溶解度合适、渗透率合适,代谢慢
大多数药呈 碱性,碱性越大,渗透率越低,溶解度先升高后降低
- 低溶解度的后果
- 生物活性低
服用量需求大
意想不到(差)的效果
昂贵的继续开发费用 - 影响药物溶解度的因素很多
- 化合物结构
溶质种类溶液组成
pH
温度
... - 如何提高溶解度
-
结构修饰:添加离子化基团、添加氢键、添加极性基团、减少分子量、空间替换,创建一个药前体
药物络合
- 转运者效应:转运者能影响药物的吸收,一般是增强
- 有些转运者能摄取药物,加强吸收;有些能使药物流出,减弱药物的吸收
- Pgp -- 糖蛋白
- p-糖蛋白大量存在于细胞屏障中,拥有保护功能
血-脑屏障,小肠,大肠,肝脏,肾脏,肾上腺,子宫 - 关于 Pgp(P-糖蛋白)流出基底的 4 原则
-
更容易成为 P-糖蛋白 基底的化合物
1、分子量 > 400
2、N + O ≥ 8
3、酸性,且 pKa > 4不容易成为 P-糖蛋白 的底物(不容易被分解流出)化合物的结构特征
1、N + O ≤ 4
2、MW < 400
3、碱性,且 pKa < 8 - 与药物有关的化合物的性质的设计
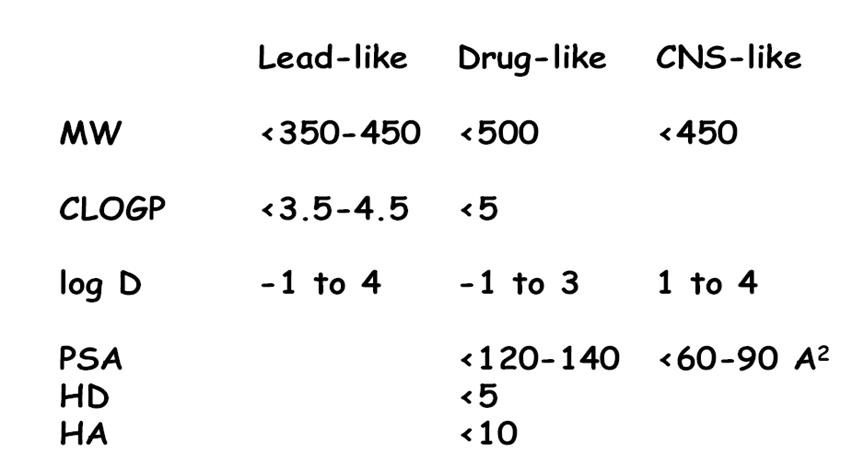
- ADME
- "ADME" 即 "毒药物动力学",指机体对外源化学物的吸收(absorption)、分布(distribution)、代谢(metabolism)及排泄(excretion)过程。
- ADME 的必要性
-
功效
安全性注射的药物对溶解度的要求更高
要失败就越早失败越好
- 药物开发早期的 ADME 评定
-
类药体的电脑模拟
实验测试
查阅物理化学性质手册
体外测试所有的 ADME 性质最终都需要用一起测量
- 提高药物与靶标作用效果的手段
- 更换取代基
结构改变
扩展/收缩碳链
扩展/收缩环
环变更
化合物基团的刚性化
柔性化
生物电子等价性转换
简化结构
氧化 - 替换取代基
- 烷基替换,能改变结合强度,改善选择性
芳香替换 - 扩展药物结构
- 可能更有利于药物与受体结合,例如 ACE inhibitors(血管紧张素转换酶抑制剂)
- 药物候选化合物判据
- 重点研究药物与靶的作用位点
服药方式的生物利用度
作用的持续性
药物在作用位点的分布情况(例如:如何穿越血脑屏障) - 药物候选化合物的确立
- 先导化合物优化
因公司而异 - 药物公司的标准
- 不同的公司不同,不同的药剂不同
- 制剂要素
- 1、静脉注射:要求较高的溶解度
2、口服:溶解度也不能过低(低于 10μg/mL)
3、盐性与晶体性质 - 药物/化合物专利
- 1、化合物必须是新颖的
2、守住秘密
3、专利的有效期是:20年,药物一经上市,专利保护则减少 6-10 年,可延期,但是不能过久,专利保护费高昂
RAS - Renin-Angiotensin System
药物对 RAS 的影响;RAS:肾素-血管紧张素系统
- RAS 系统
-
控制着动脉血压、体液电离平衡
两部分:肾素、血管紧张素转换酶
这两种酶控制着 血管紧张素肽原 向 血管紧张肽2 的转换
血管紧张肽2 是有效的血管收缩剂,影响着 肾功能 与 心血管系统所以,RAS 系统的调控对于心血管疾病的治疗与预防有着重大的作用
- RAS 系统的主要组成
- 血管紧张肽原
肾素
血管紧张素转换酶(使血管紧张素肽原向血管紧张素转换)
血管紧张素1
血管紧张素2(主要作用)
血管紧张素3
 妨碍 血管紧张素2 的合成与抑制 血管紧张素2 与其受体的结合,意味着我们能够我们能够对很多心血管疾病进行预防和治疗
妨碍 血管紧张素2 的合成与抑制 血管紧张素2 与其受体的结合,意味着我们能够我们能够对很多心血管疾病进行预防和治疗
RAS 与 血压升高有着密切的联系
RAAS - 肾素-血管紧张素-醛固酮系统
- ACE 的作用后果
- 导致 血管紧张素2 带来的 高血压
导致大量产生全部通
使我们的抗高血压的血管扩张药物失效
血管紧张素转换酶抑制剂(ACE Inhibition)的 受体(靶标)是 血管紧张素1,能阻止 血管紧张素1 向 血管紧张素2 的转换
肾素抑制剂 能抑制 血管紧张肽原 向 血管紧张素1 的转换
在设计心血管疾病(高血压)药物时,一个重要的靶标就是 ACE -- 血管紧张素转换酶
- ACEIs -- 血管紧张素转换酶抑制剂的分类与差异
-
在美国(FDA)批准使用的有 12 种
所有这些 ACEIs 原理相同:抑制 血管紧张素1 向 血管紧张素2 的转换这些药物存在的差别是
1、药效
2、起作用的是药物本身,还是前体药物转换后的产物起作用
3、代谢产物
4、动力学特性 - ACEIs 在治疗心血管疾病的常见副作用
- 干咳
血管性水肿
再生障碍性贫血
结膜炎
头痛
... - 血管紧张素2(ANG 2)的受体 AT1 带来的主要作用
- 血管收缩(直接收缩、增强去甲状腺素的作用、增加交感神经放电)
盐滞留(增加 Na+ 吸收、使肾上腺皮质合成释放醛固酮、肾血管收缩)
血管畸形生长与肥大
血管紧张素2 是器官受损的主要罪人
- 血管紧张素2 受体 AT1 的拮抗剂
- 早期基于 天然缩氨酸 的 肌丙抗增压素
之后是非基于天然缩氨酸的 洛沙坦
之后就是 依普沙坦 - 第一种非缩氨酸 AT1 拮抗剂 -- 洛沙坦
- 口服
持续有效
所有 AT1 选择性拮抗剂的原型
用于验证 缩氨酸药物分子 或 ACEIs 的有效性
大约 14% 的氯沙坦会被 CYP2C9 等酶氧化
氯沙坦不良反应:CNS System 影响
- AT2 为 ANG2(血管紧张素2)带来的作用
- 与 AP1 相似
- 找到一个 AT2 拮抗剂的必要性
- 抵抗癌症、创伤愈合、心里衰竭、肠道易激综合症
少数 AT2 受体拮抗剂全部是 缩氨酸 类化合物
- AT1 与 AT2 在结构上
- AT2 363 a.a AT1 359 a.a
AT1 与 AT2 在蛋白质水平上 34% 相似度
AT1 与 AT2 都与 ANG2 结合 - 为什么不使用四氮唑
-
因为优缺点
合成问题
代谢问题 - L-162,313 是 AT1 和 AT2 的双重拮抗剂
- 存在的缺陷:选择性低或者活性低
- P450抑制剂
- P450 是部分屏障能达到阻拦药物的主要工具
抗肿瘤药物
- 肿瘤三要素
- 遗传
致癌物质
引发剂、引发因素 - 化疗
- 始于:1940s,使用 氮芥子气 和 抗叶酸制剂
- 靶向治疗 革命已经到来
- 癌症的分子靶在大型的研究项目中被发现
药物研究技术的集成应用 - 抗肿瘤药物中的成功与局限
- 细胞毒性药物治疗
新的 基于机制 的抗肿瘤分子的发现
靶向药物、抗体、小分子 被发现 - 典型抗癌的抗体和小分子
- 郝塞丁
伊马替尼
等 - 抗癌抗体与小分子的副作用
- 心力衰竭
间质性肺炎
对胎儿造成影响(孕妇服用的话) - 白血病临床症状
- 疲劳,减重,白血球数目超过 50000/μL
- 白血病病因与基于机制的药物
- 9 号染色体异常
Bcr-ab1基因和融合蛋白酪氨酸激酶(PTKs)是病变细胞赖以生存的一种酶,可以从这里下手
Bcr-ab1 作为白血病的治疗靶标 - 在格列卫之前的疗法
- α干扰素或白消安等药物
肝脏X-ray疗法
配对骨髓移植 - PTKs(蛋白酪氨酸激酶) -- 控制细胞的增值和扩散
- PTKs 在细胞的分化、增值中扮演重要角色
每个细胞,200-300 中蛋白激酶
蛋白激酶与肿瘤有着密切的关系 - ATP-定向抑制剂 与 基于底物的抑制剂
- 底物抑制剂被认为是更好的选择
ATP-定向抑制剂的实现也更难
五个表皮生长因子受体酪氨酸激酶抑制剂正在使用中
糖尿病
- 三种糖尿病
- Type 1
Type 2
妊娠期糖尿病 - 糖尿病并发症
- 中风
心脏病
肾病
眼病
神经损伤 - 血糖
-
在中枢神经系统中
1、大脑使用葡萄糖作为基本供能物质
2、大脑不能储存/产生葡萄糖在中枢神经系统之外
1、糖原
2、甘油三脂
α-胰岛细胞 制作 胰高血糖素
β-胰岛细胞 制作 胰岛素
血糖过高,胰岛素进入 肝脏细胞、肌肉细胞、脂肪细胞 存储 糖、脂肪、蛋白质,以降低血糖
- 血糖高带来的影响
- 混混欲睡
口渴
心情激动
血糖过低时,胰高血糖素进入肝脏细胞,将糖原分解,转为葡萄糖使血糖升高
- 血糖低的影响
- 虚弱
出汗
焦虑、难以集中注意力 - Type 1 糖尿病
- 产生胰岛素的细胞受损
导致胰岛素依赖
30岁前能被侦测 - Type 2 糖尿病
- 生成的胰岛素缺乏
胰岛素作用力不够
一般 40 岁之后被侦测
可能导致b细胞受损
