@zhoujj2013
2017-08-25T15:50:19.000000Z
字数 3754
阅读 2632
XXX RNA-seq信息分析结题报告(基础分析)
report
生物信息分析流程
对于无参考基因组的转录组分析,可先将测序所得的序列拼接成转录本,以转录本为参考序列,进行后续分析。信息分析流程图如下:
数据质量控制
1. 测序数据质量分布
如果测序错误率用e表示, Illumina HiSeq 2000/Miseq的碱基质量值用表示,则有:。Illumina Casava 1.8版本碱基识别与Phred分值之间的简明对应关系见下表:
| Phred分值 | 不正确的碱基识别 | 碱基正确识别率 | Q-score |
|---|---|---|---|
| 10 | 1/10 | 90% | Q10 |
| 20 | 1/100 | 99% | Q20 |
| 30 | 1/1000 | 99.9% | Q30 |
| 40 | 1/10000 | 99.99% | Q40 |
对于第二代测序技术,测序错误率分布具有两个特点,具体见图 1:
(1) 测序错误率会随着测序序列(Sequenced Reads)的长度的增加而升高,这是由于测序过程中化学试剂的消耗而导致的,并且为Illumina高通量测序平台都具有的特征。
(2) 前6个碱基的位置也会发生较高的测序错误率,而这个长度也正好等于在RNA-seq建库过程中反转录所需要的随机引物的 长度。所以推测这部分碱基的测序错误率较高的原因为随机引物和RNA模版的不完全结合所致。一般情况下,单个碱基位置的测序错误率应该低于1%。(只适用于RNAseq)
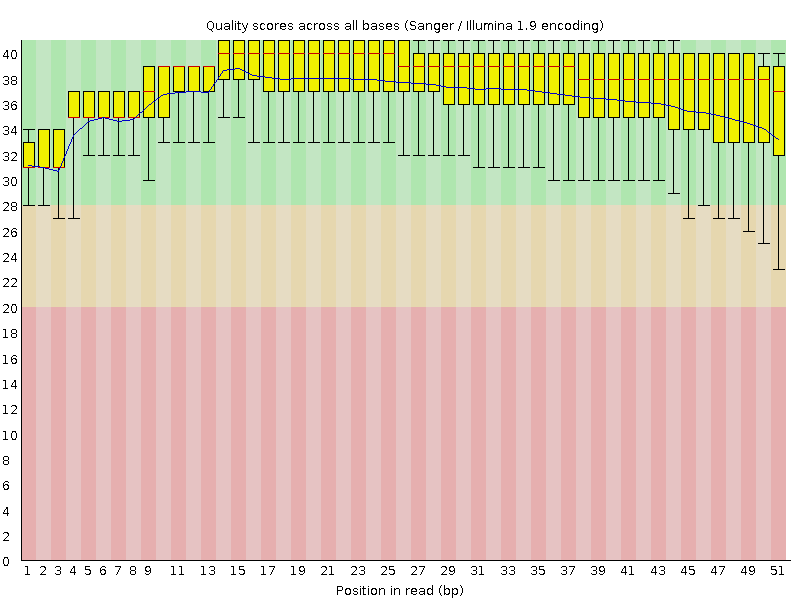
图1 测序错误率分布图
横坐标为reads的碱基位置, 纵坐标为单碱基错误率
2. clean fastq统计数据
样品测序产出数据质量评估情况详见表1。
表1 数据产出质量情况一览表
| Sample | Raw Reads | Clean Paired reads | Clean reads | Filtered reads | Clean reads(%) | GC(%) |
|---|---|---|---|---|---|---|
| Con_1 | 7448520 | NA | 7425666 | 22854 | 31 | 36 |
| Con_2 | 7448520 | NA | 7425666 | 22854 | 31 | 36 |
Sample: 样品名。
Raw reads: 统计原始序列数据,以四行为一个单位,统计每个文件的测序序列的个数。
Clean Paired reads: 达到质量标准的Paried-end测序片段。
Clean reads: 计算方法同 Raw Reads, 只是统计的文件为过滤后的测序数据。后续的生物信息分析都是基于Clean reads。
Filtered reads: 未能达到质量标准的测序序列个数。
Clean reads (%): 达到质量标准测序数据的百分比。
GC content: 计算碱基G和C的数量总和占总的碱基数量的百分比。
3. 重复reads统计 (DNA会省去此部分,例如:ChIP-seq, Exome-seq, 重测序)
样品测序产出数据重复测序片段评估情况详见表2。
表2 重复测序片段情况一览表
| Sample | Clean reads | Dup. reads | Uniq. reads | Dup. rate(%) |
|---|---|---|---|---|
| Con_1 | 7448520 | 1425666 | 6022854 | 19.1 |
| Con_2 | 7448520 | 1425666 | 6022854 | 19.1 |
Sample: 样品名。
Clean reads: 统计质量过滤后序列数据, 以四行为一个单位,统计每个文件的测序序列的个数。
Dup. reads: 在测序数据中的,来自PCR及测序过程optical的重复序列,这部分序列是人工造成的,不会用到后续分析。
Uniq. reads: 在测序文库中,序列唯一的测序序列个数。
Dup. rate (%): PCR及测序过程optical的重复序列占总序列的百分比。
注意:对于双端测序(paired-end sequencing),此分析只包含过滤后双端符合质量要求的数据。
短序列比对
1. 短序列比对统计
以参考序列(ref)作为序列库,将每个样品的clean reads对ref做比对。该过程我们采用了RSEM软件,tophat2软件(RNA-seq)或者BWA, STAR软件(DNA-seq)(Li et al., 2011),RSEM, Tophat2中使用到的bowtie2参数mismatch 2,(bowtie默认参数)。比对统计结果见下表:
| sample id | clean reads | mapped reads | mapping rate(%) |
|---|---|---|---|
| s1 | 10000 | 8000 | 80% |
| s2 | 10000 | 8000 | 80% |
sample id: 样品名。
clean reads: 输入比对软件的原始序列数据,以四行为一个单位,统计每个文件的测序序列的个数(默认是过滤后数据)。
mapped reads: 比对到参考序列的测序序列个数。
mapping rate(%): 比对到参考序列的测序序列占总序列的比例。
2. 短序列比对分布
| region type | mapped reads | mapped percentage(%) |
|---|---|---|
| 3UTR | ||
| miRNA | ||
| ncRNA | ||
| TTS | ||
| pseudogenes | ||
| Exon | ||
| Intron | ||
| Intergenic | ||
| Promoter | ||
| 5UTR | ||
| snoRNA | ||
| rRNA |
Promoter: defined from -1kb to +100bp)
TTS: defined from -100 bp to +1kb)
Exon: CDS Exons
5UTR: 5' UTR Exons
3UTR: 3' UTR Exons
表达量计算
1. 基因表达量计算
采用Cufflinks软件,基于比对结果,对每一个基因进行表达量计算。同样采用Cufflinks软件对每一个基因的亚型进行表达量计算。
| 基因名字 | 表达量(FPKM/RPKM) |
|---|---|
| MALAT1 | 10 |
| MYOD1 | 30 |
| MYOG | 90 |
2. 转录本表达量计算
采用Cufflinks软件,基于比对结果,对每一个基因进行表达量计算。同样采用Cufflinks软件对每一个基因的亚型进行表达量计算。
| 基因名字 | 表达量(FPKM/RPKM) |
|---|---|
| MALAT1_trans_id1 | 10 |
| MYOD1_trans_id2 | 30 |
| MYOG_trans_id3 | 90 |
RNA-seq整体质量评估(多样品分析)
把多个样品放在一起,结合表型信息,检查基因表达是否与表型一致,可以依据些结果,选取样品进行后续高级生物信息学分析。
1. 多样品表达量
把多样品放在同一个txt文件中,有利于用R, excel等统计软件进行分析。格式如下:
| GeneID | sample1 | sample2 | ... | sampleN |
|---|---|---|---|---|
| gene1 | 5 | 9.2 | ... | XXX |
| gene2 | 5 | 9.2 | ... | XXX |
| gene3 | 5 | 9.2 | ... | XXX |
| ... | 5 | 9.2 | ... | XXX |
| geneN | 5 | 9.2 | ... | XXX |
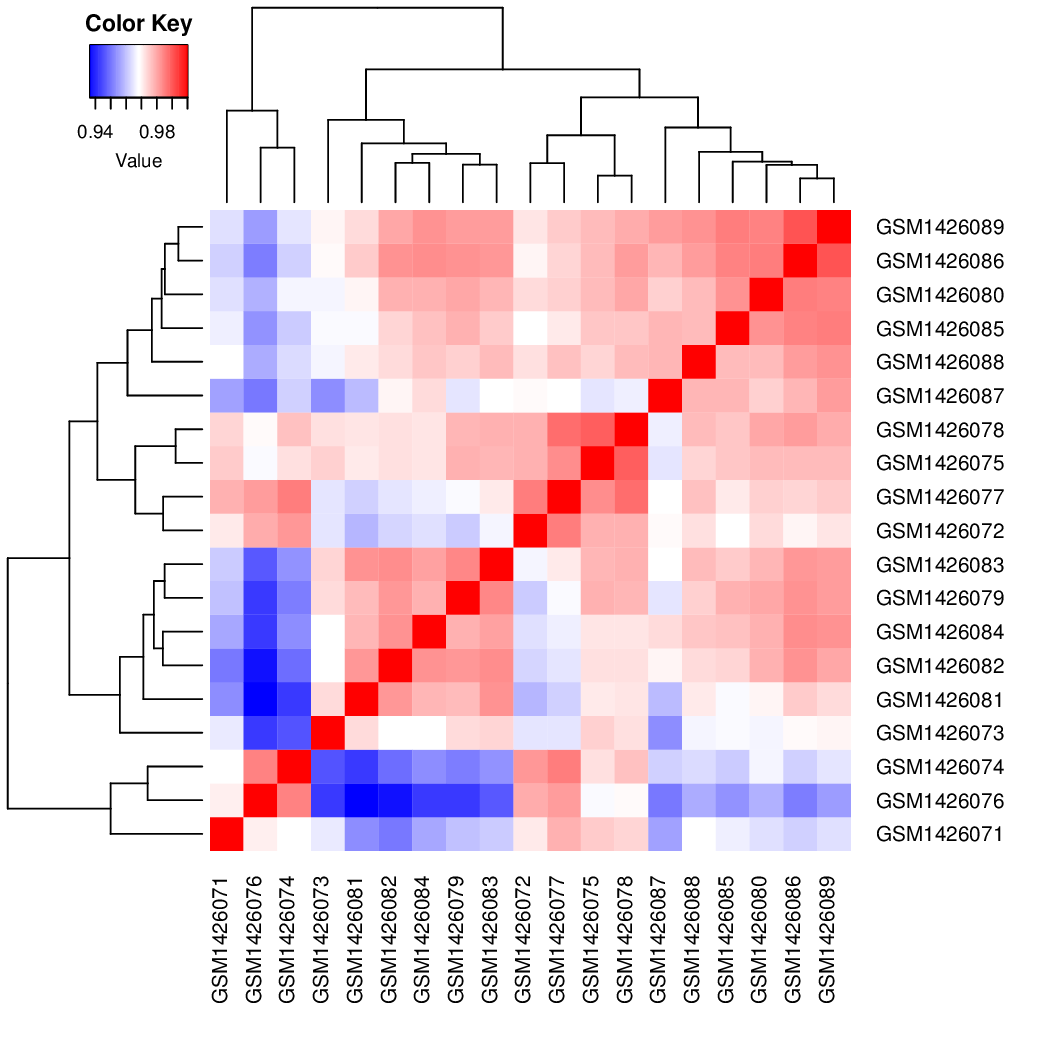
2. 样品间相关性分析
用pearson方法计算两两样品间的相关系数,用层次聚类分析对样品进行聚类。具有相似表达谱的样品会聚集在一起,聚集的每一个类里的样品具有相关的表达谱。如下图所示:
其它文件
过滤后的测序数据:
XXX.trimed.rmdup.r1.fq
XXX.trimed.rmdup.r2.fq
XXX.fastqc
比对结果文件:
xxx.acceptedhits.bam
表达量计算结果:
XXX.gene.rpkm
XXX.transcript.rpkm
多样品表达量:
xxx.merged.expr
correlation_clustering_graph.png
correlation_clustering_graph.pdf
参考文献
- http://bonsai.hgc.jp/~mdehoon/software/cluster/software.htm
- Trapnell, Cole, et al. "Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks." Nature protocols 7.3 (2012): 562.
- Team, R. Core. "R language definition." Vienna, Austria: R foundation for statistical computing (2000).
- Langmead B, Salzberg S. Fast gapped-read alignment with Bowtie 2. Nature Methods. 2012, 9:357-359.
- Bolger A M, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data[J]. Bioinformatics, 2014, 30(15): 2114-2120.
- Andrews S. FastQC: a quality control tool for high throughput sequence data[J]. 2010.
