@agpwhy
2021-11-03T14:44:32.000000Z
字数 1261
阅读 438
王胖的生信笔记第24期:下载芯片数据并简单质控
上次提到了怎么转换基因芯片里的一些id到基因名,但其实并不需要这么复杂。有更好的解决方案。感恩各位R工具包的内容贡献者。感谢健明老师的教程。
就按照上次那个例子投石问路一下吧。
设置工作环境
library(GEOquery)
library(limma)
library(umap)
Sys.setenv("VROOM_CONNECTION_SIZE" = 131072 * 2)
这里涉及到一个VROOM的链接容量设置,具体我记不得了。
setwd("/Users/wangshengming/Desktop/生信笔记/第二十四期")
搞一搞矩阵
gset <- getGEO("GSE158850", GSEMatrix =TRUE, getGPL=FALSE)
if (length(gset) > 1) idx <- grep("GPL13497", attr(gset, "names")) else idx <- 1
这里的GPL13497是芯片名称,对于这个数据其实就是1,还好。
gset <- gset[[idx]]
ex <- exprs(gset)
对于矩阵做一个log2转换
qx <- as.numeric(quantile(ex, c(0., 0.25, 0.5, 0.75, 0.99, 1.0), na.rm=T))
LogC <- (qx[5] > 100) ||
(qx[6]-qx[1] > 50 && qx[2] > 0)
if (LogC) { ex[which(ex <= 0)] <- NaN
ex <- log2(ex) }
作图看一下各个样本基本情况。整体来说各个指标log处理后,基本都比较均一。
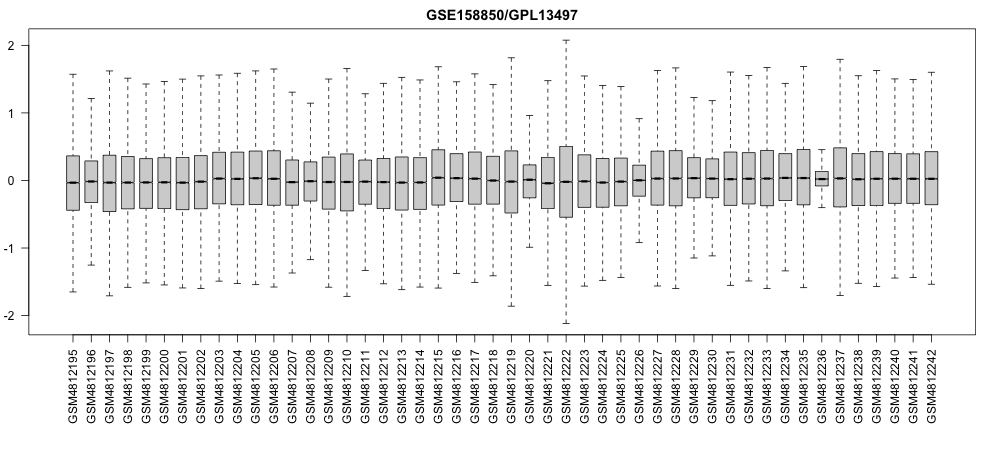
换一种方式看一下。
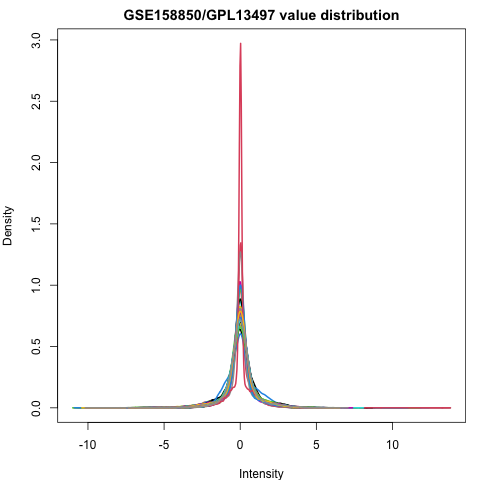
整体来说数据还是可以的。不过还是可以处理一下。
其实还有一些其他指标可以看,比如标准差(variance),umap图等。
再处理一下
然后不同批次之间一般会有一定差异,这些差异可能由各种原因造成(比如不同时间机器精度不同,不同批次环境不同等)。这些可以认为是批次效应(Batch Effect),可以使用limma包内置的函数矫正。
ex=normalizeBetweenArrays(ex)
然后再看下boxplot
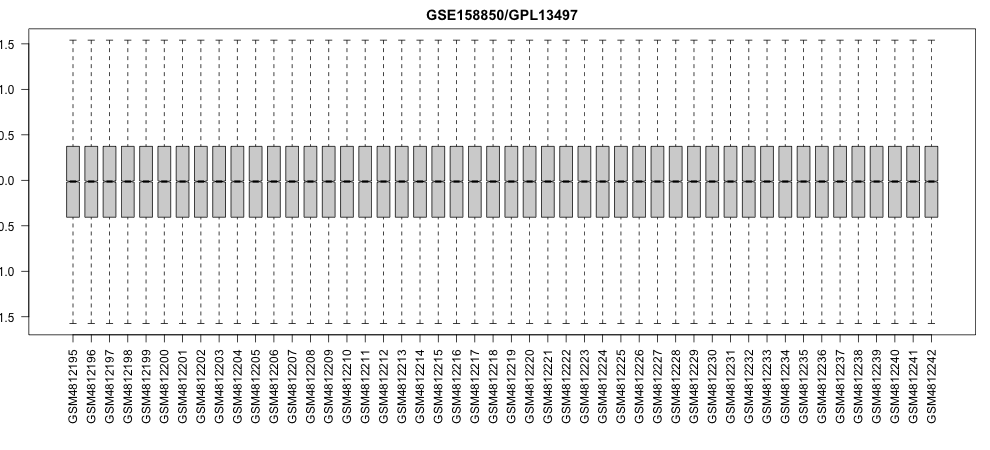
boxplot(ex, boxwex=0.7, notch=T, main=title, outline=FALSE, las=2)
果然很好了。
最后匹配一下ID和基因名
这里和上一次是类似的,不过下一次做后续分析的时候,教给大家一个更简单的方式。
library(tidyverse)
ex <- as.data.frame(ex)
ex <- ex %>%
mutate(probe_id=rownames(ex)) %>%
inner_join(ids,by="probe_id") %>%
select(probe_id, symbol, everything())
ex <- ex[!duplicated(ex$symbol),]
rownames(ex) <- ex$symbol
ex <- ex[,-(1:2)]
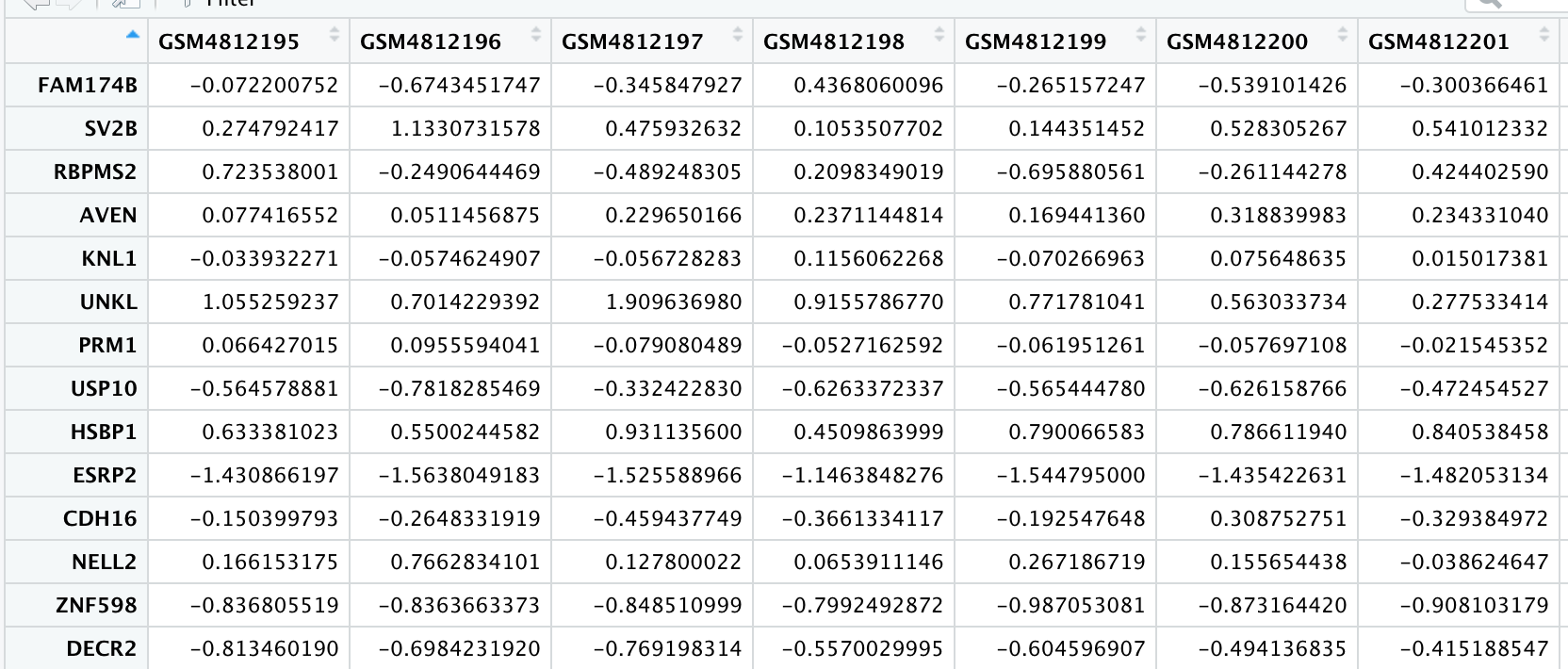
已经有这些了。可以进行后续分析了。
后面一期给大家推荐个傻瓜工具包,给大家更好的体验。
