@agpwhy
2022-01-18T02:47:33.000000Z
字数 2599
阅读 598
王胖的生信笔记第34期:单细胞树型图
事情的起因
一周多前,小老板给我丢了这么一篇我之前丢给过他看的文章 Lineage tracking reveals dynamic relationships of T cells in colorectal cancer (doi:10.1038/s41586-018-0694-x)。里面当初比较感兴趣的是他关于T细胞的一些细节描述。但那天他在翻的时候,看到了这么一张图。
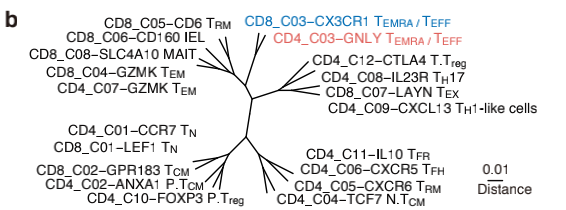
问我能不能做一下类似的

然后一研究就发现其实之前有做过类似的(详见我笔记第13期)。但是这篇文章用的计算各群之间距离的方法比较细:Similarities of the signature gene expressions of T cell clusters. Distance: (1-Pearson correlation coefficient)/2。解决这个问题就解决了一段时间。现无偿公布如下。
找个示范数据
原文的数据太大了,就找个替代的数据。
library(SeuratData)library(Seurat)library(tidyverse)data("bmcite")bmcite$celltype.l2 <- as.factor(bmcite$celltype.l2)
这是一个30k Bone Marrow Cells的参考数据集。里面已经进行了细胞类型鉴定和注释,共分了27群,非常适合进行示范。不会搞这个参考数据集的话,下次我来搞个视频试试?
选出代表基因计算矩阵
对于计算相似性,拿所有基因去算太多,就挑选一些代表性的基因。
df <- data.frame()for(i in 1:length(levels(bmcite$celltype.l2))) {df_i <-FindMarkers(bmcite,ident.1 = levels(bmcite$celltype.l2)[i],group.by = "celltype.l2",logfc.threshold = 0.25,only.pos = T,)df_i$cluster <- levels(bmcite$celltype.l2)[i]df_i$gene <- rownames(df_i)df <- rbind(df, df_i)}top=10TopMarker1 <- df %>% group_by(cluster) %>%top_n(n = top, wt = avg_log2FC)genelist <- unique(TopMarker1$gene)
然后根据筛选的的基因构建数据矩阵
dat <- AverageExpression(bmcite,assay="RNA",features = genelist,group.by = "celltype.l2")
后面我的操作就有点丑陋了。
计算距离
Distance: (1-Pearson correlation coefficient)/2。就文中短短几句话,自己想要想破头。
最后反正我的这个实现方式挺丑陋的,就当抛砖引玉吧。
mat <- dat[[1]]mat <- t(mat)df <- dat[[1]]df <- as.data.frame(df)dim(df)z <- matrix(NA,27,27)for (i in 1:27){for (j in 1:27){a = cor(df[,i],df[,j],method="pearson")z[i,j]=(1-a)/2}}colnames(z) <- colnames(df)rownames(z) <- colnames(df)test <- ztest <- test[-1,]for (i in 1:26){for (j in (i+1):27){test[i,j]= NA}}sf <- dist(mat,method = "euclidean")number <- numeric()for (i in 1:26){number[i] <- (((26+27-i+1)*(i-1))/2)}for (i in 1:26){for (j in i:26){sf[(((26+27-i+1)*(i-1))/2)+j+1-i]= test[j,i]}}mydatatest<-hclust(sf)
最后就是作图了
ggtree(mydatatest) + geom_tiplab(hjust = 0, size = 3) + xlim(0,.3) + ggtitle("Zhang's Paper") +theme(title = element_text(size = 24,face = "bold", hjust = .5))
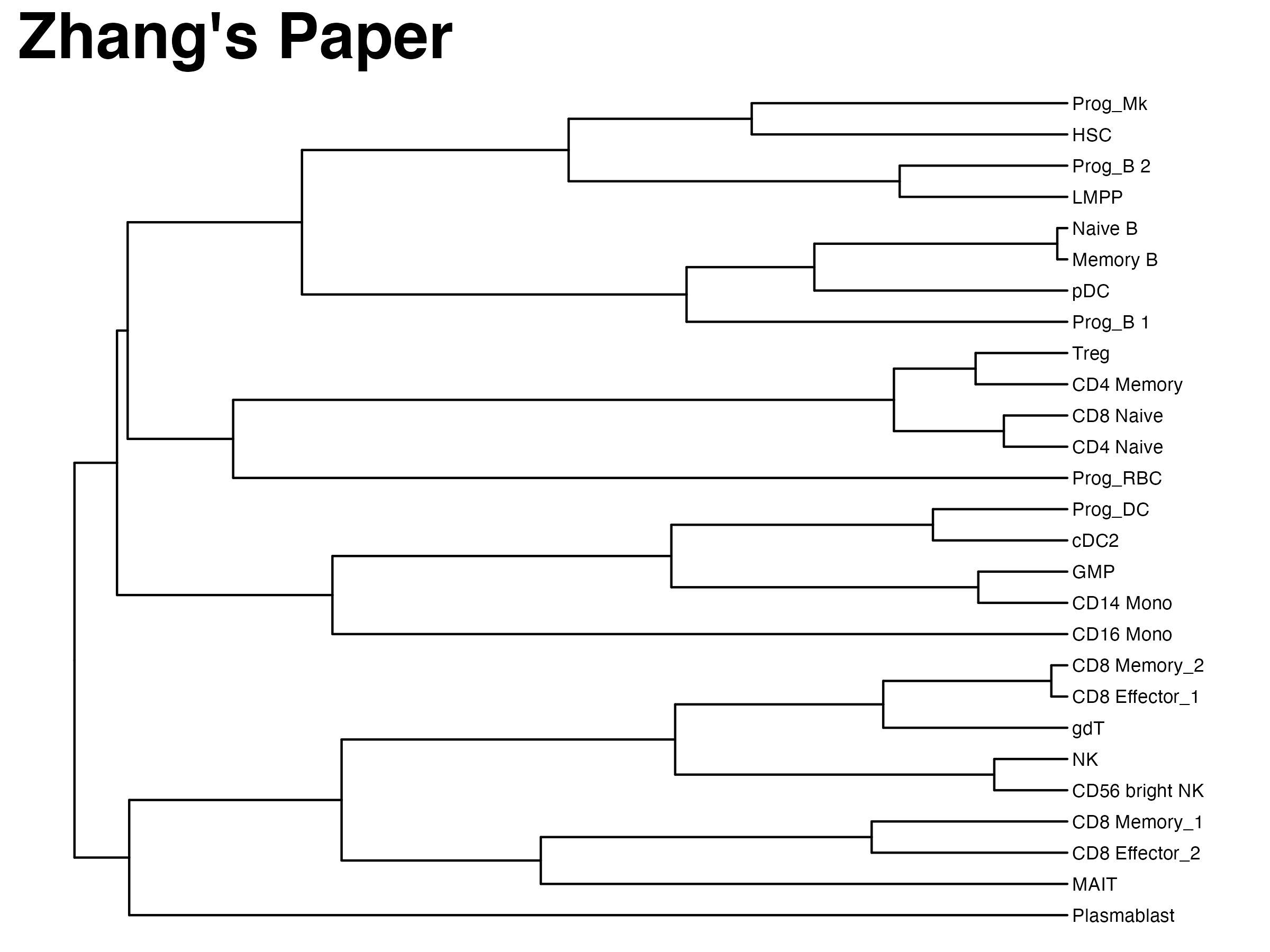
这就是最基本的树形图结构了。如果要改成他文章中的那种展示类型,可以看看ggtree的说明书(https://yulab-smu.top/treedata-book/chapter4.html)。
看着张泽民老师那篇文章里用的是daylight这种分布方式。
ggtree(mydatatest, layout = "daylight")+geom_tiplab(hjust=0,size=4)+ggtitle("Zhang's Paper")+theme(title = element_text(size = 24,face = "bold", hjust = .5))
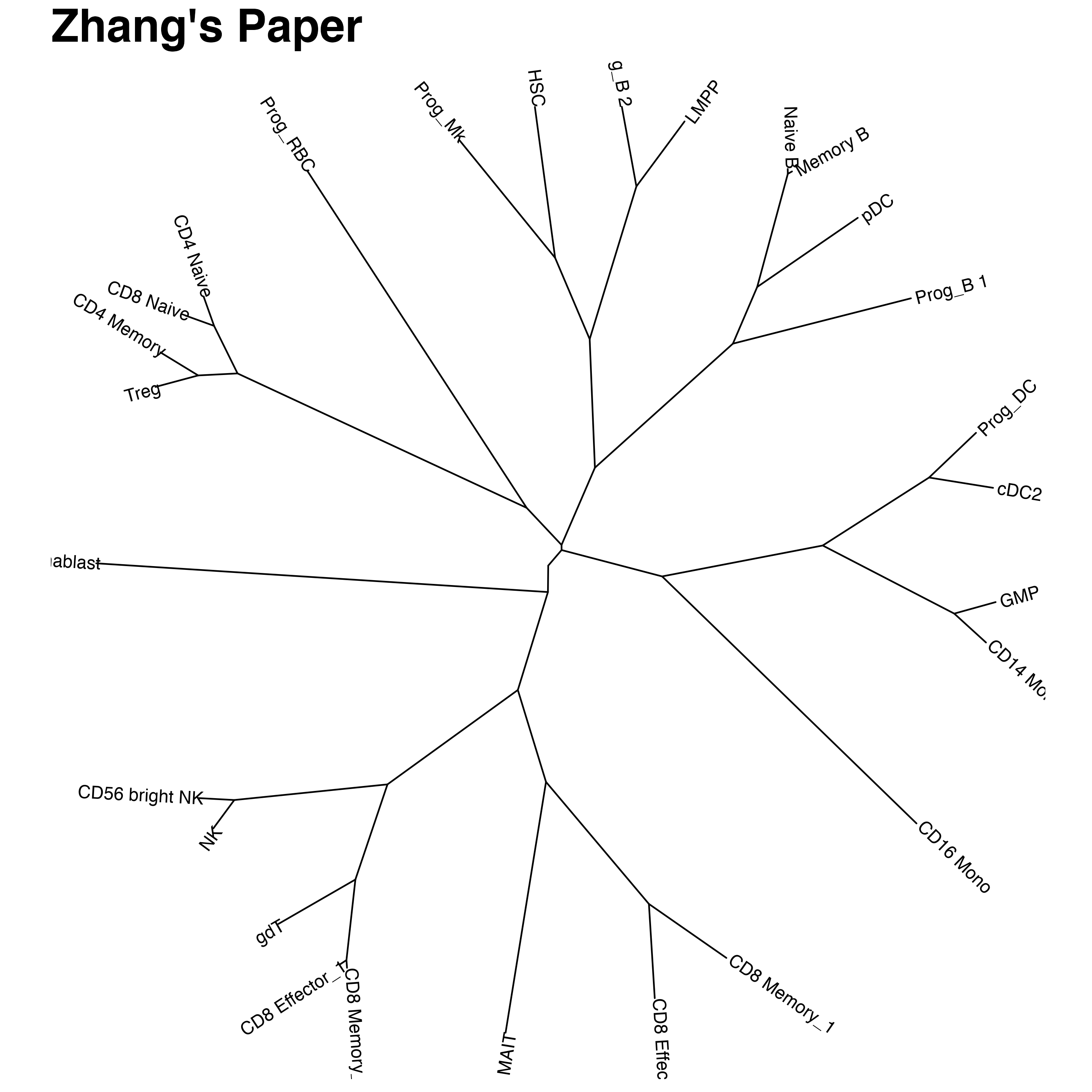
其他各种计算距离方式

R里面还自带这些计算距离方式,对于数学我很外行。可能过要看哪种更符合你的预期了。
mydata<-hclust(dist(mat,method = "euclidean"))ggtree(mydata)+geom_tiplab(hjust= 0,size=3)+xlim(0, 1500)+ggtitle("Euclidean")+theme(title = element_text(size = 24,face = "bold", hjust = .5))ggtree(mydata, layout = "daylight")+geom_tiplab(hjust = 0,size=4)+ggtitle("Euclidean")+theme(title = element_text(size = 24,face = "bold", hjust = .5))
这是欧几里得计算距离方式
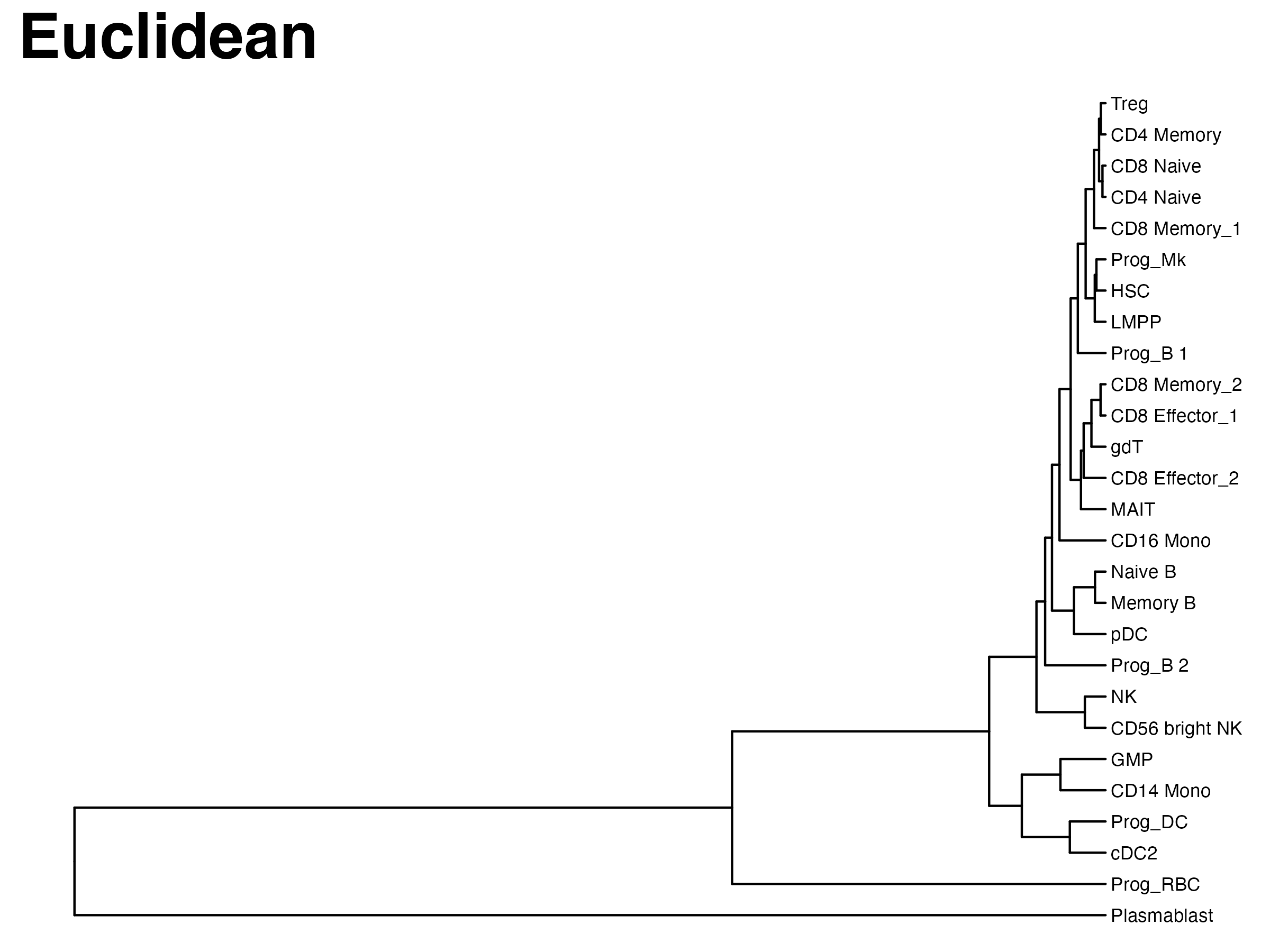
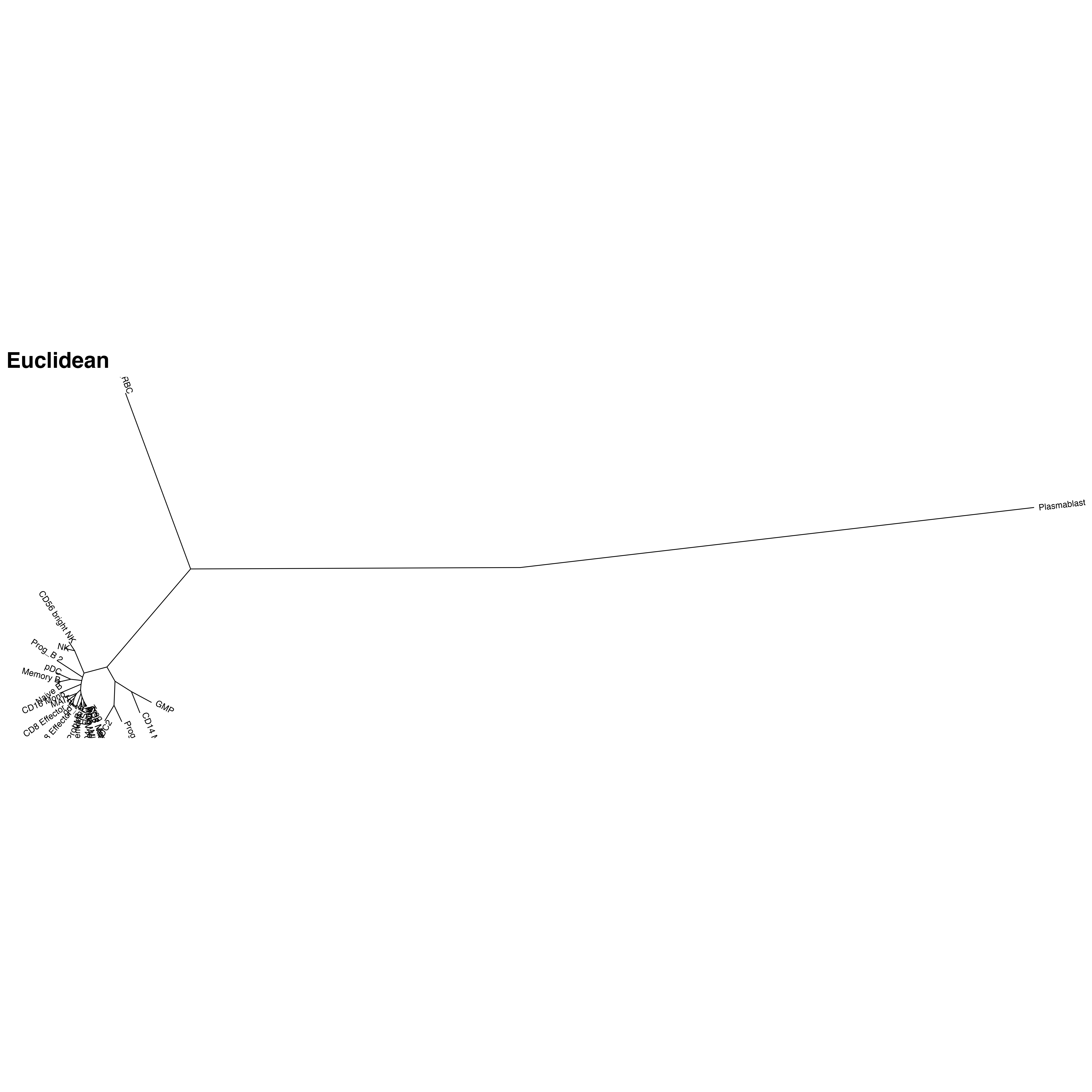
很显然展示的结果不太一样。血小板,红细胞和其他骨髓细胞区别非常大,这可能反而是更贴近实际的情况吧。不知道张老师为什么选用了他那种方式。
其他的计算距离结果:
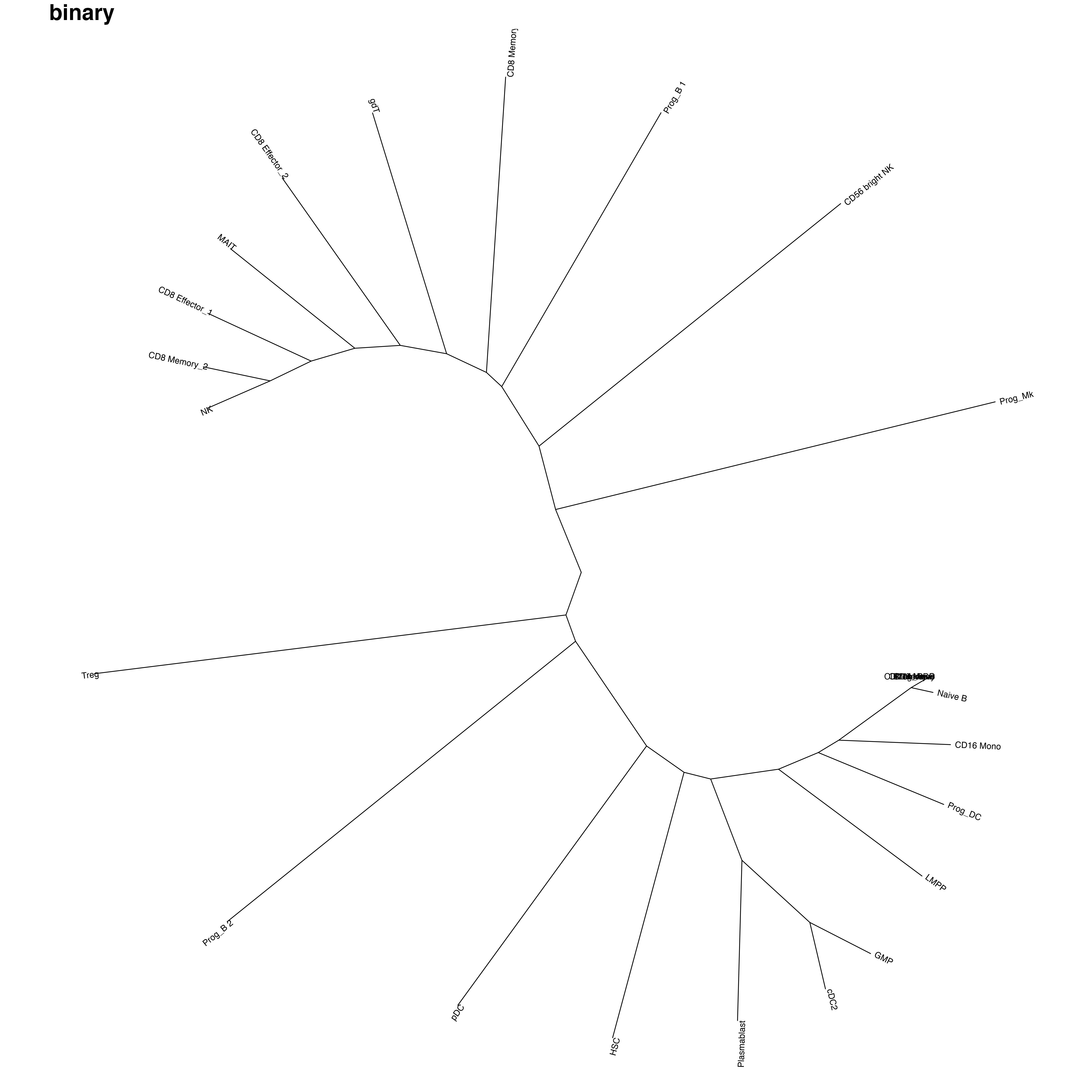
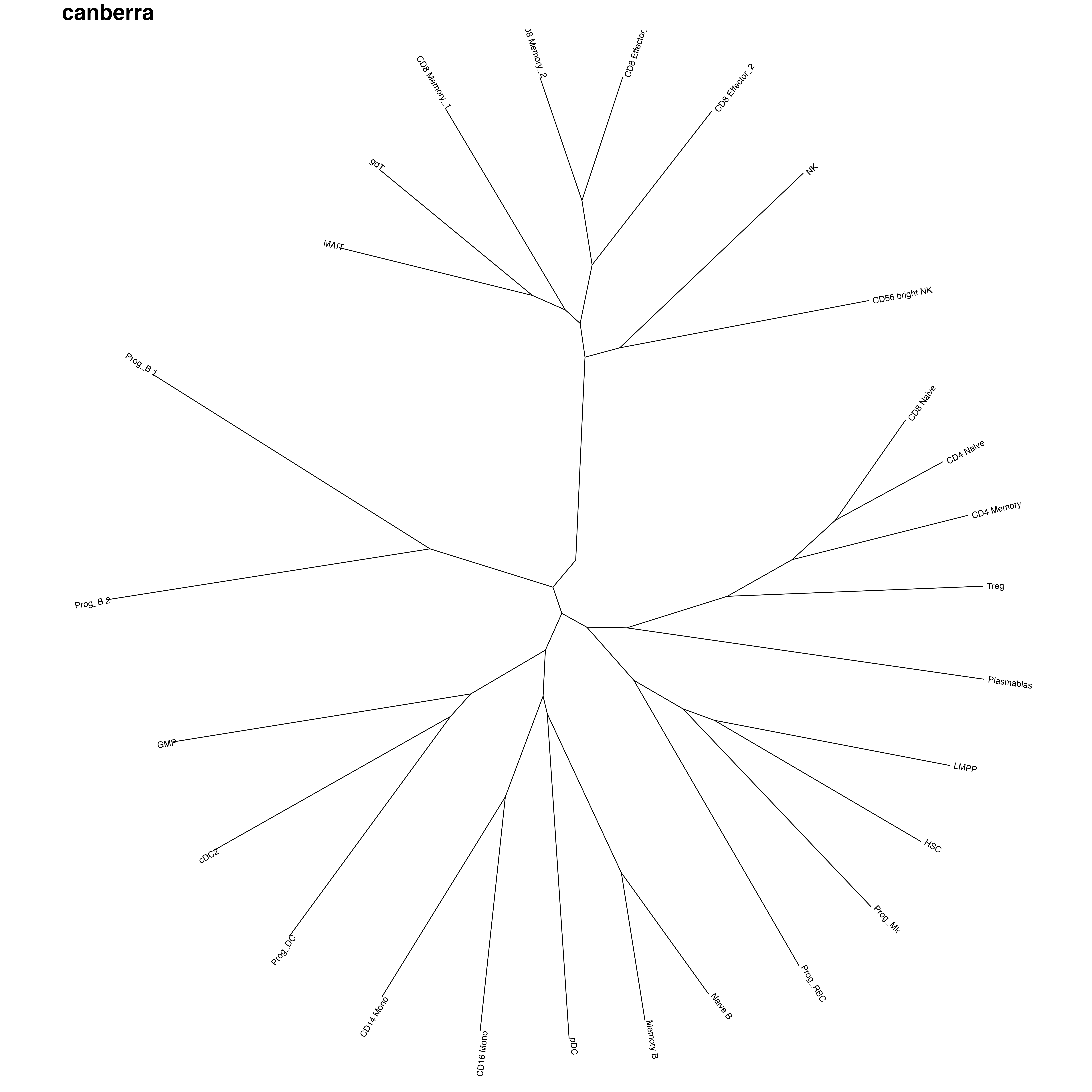
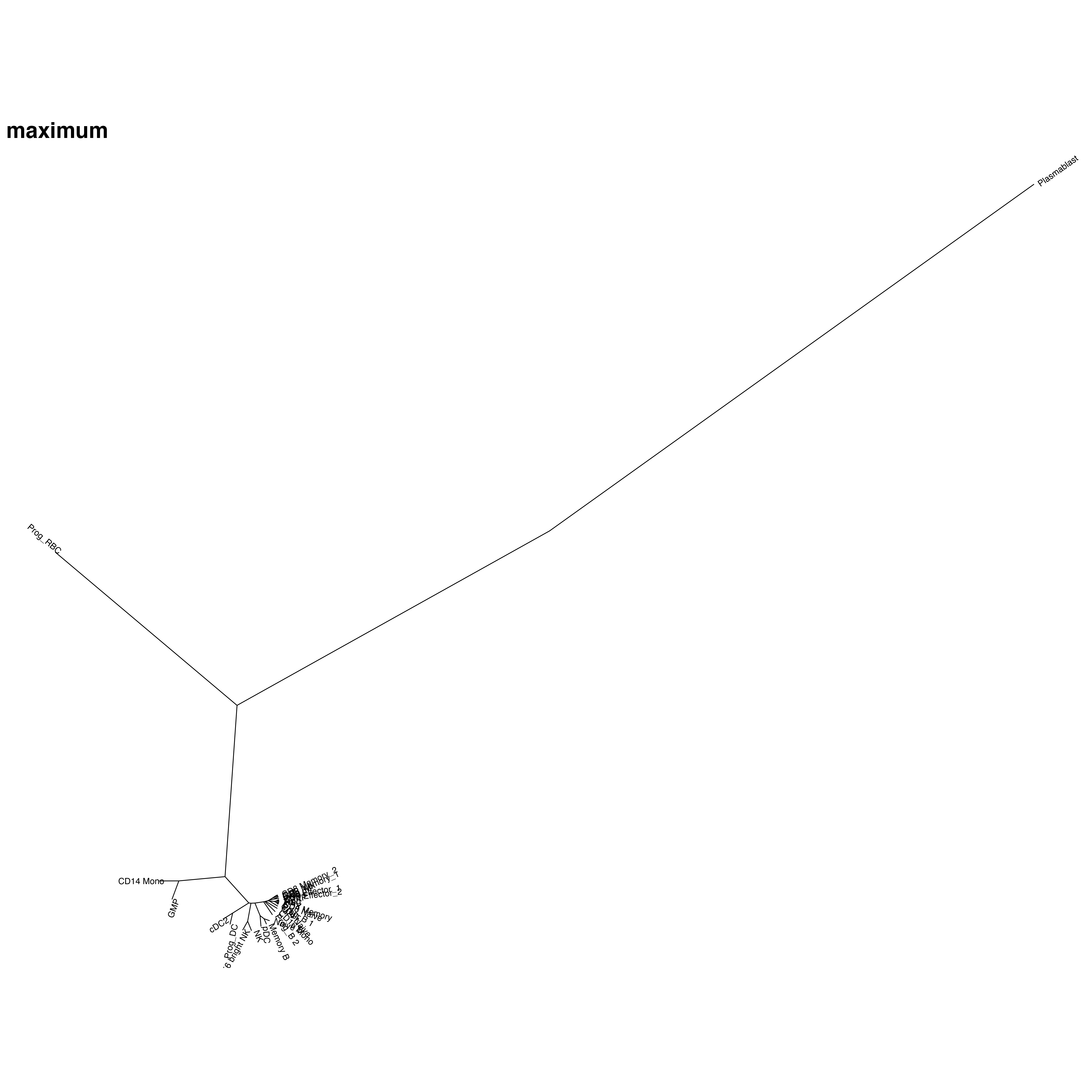
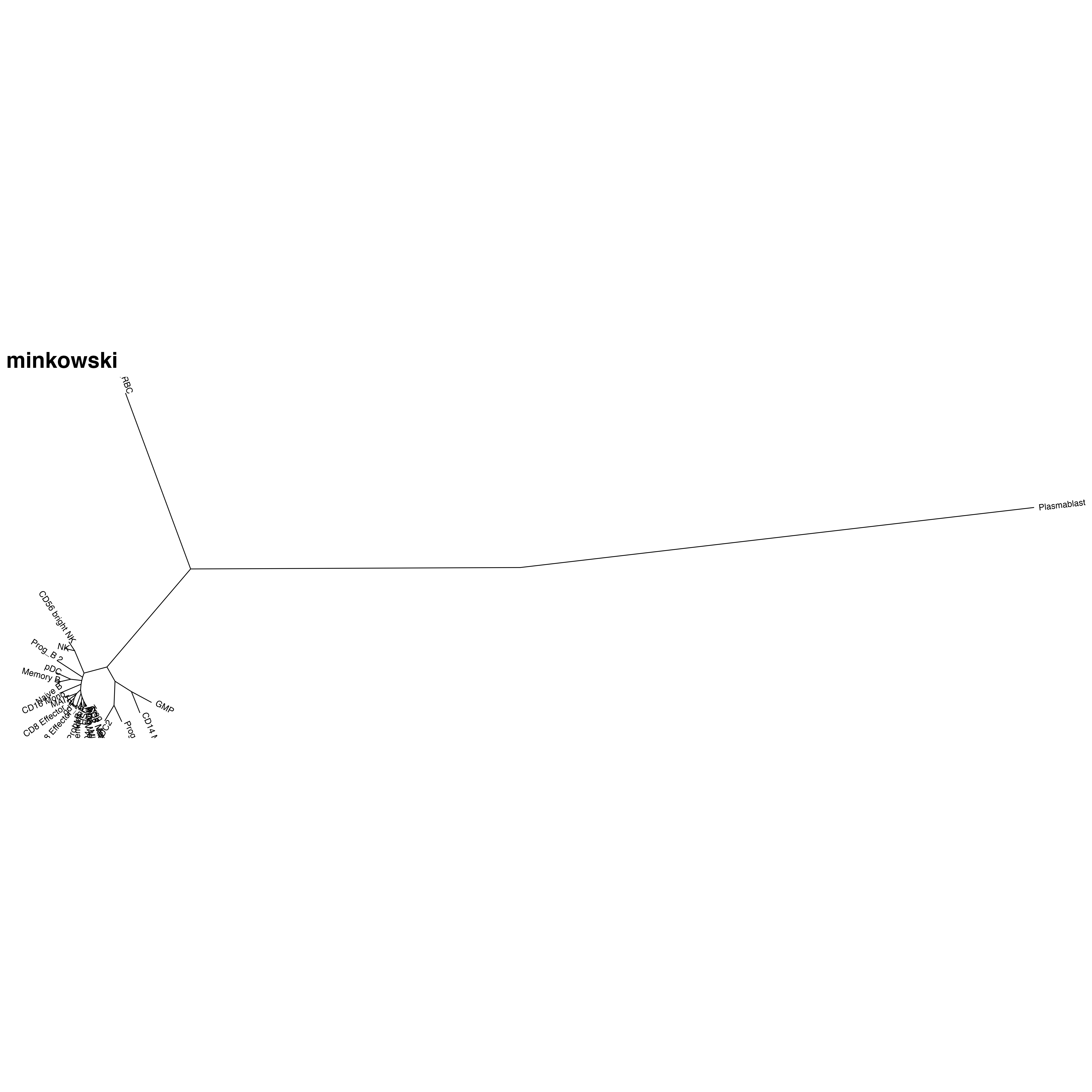
【minkowski方法结果和euclidean非常类似,不知道是不是什么数学上的原因】
