@agpwhy
2022-05-30T12:43:11.000000Z
字数 3032
阅读 683
生信文章代码复现可能踩哪些坑?-王胖的生信笔记第44期
对于学习生物信息学,很重要的一点就是要学会使用复制黏贴。关于这次分享什么,我没有任何想法,就想挑战一下随便搜一个给出代码和数据的文章做一下复现的尝试。复现嘛,最重要的就是做好复制黏贴的工作。
复制黏贴的难度
复制黏贴有什么难度啊?你可能有这样的疑问。之前我曾经在笔记里写下过这样一段暴论:“学会打字,就会了R的20%;学会复制黏贴,就又会了R的20%;如果还会灵活机动地使用搜索引擎的话,就再会了R的20%”。能够保证你掌握20%的R,这个复制黏贴还是有一定门道的。
今天要复现的内容来自这篇文章《Bioinformatics Analysis of Metabolomics Data Unveils Association of Metabolic Signatures with Methylation in Breast Cancer》。文章具体的细节就不说了,就简单的说一下里面的分析思路:
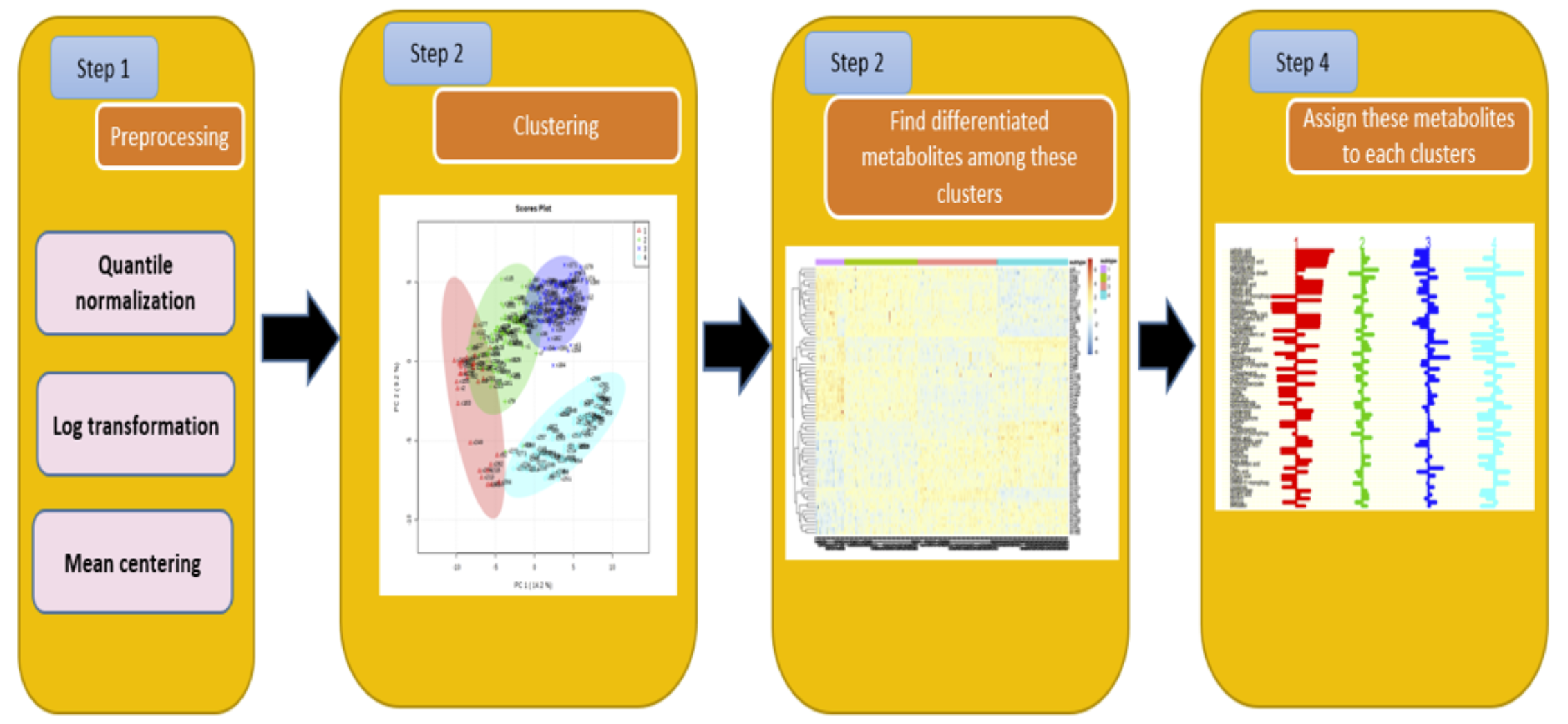
首先收集代谢组学数据,通过预处理后根据不同方法分出样本的Cluster,然后在不同Cluster间计算代谢物的差异,以及甲基化的差异。同时将部分结果结合其他公共数据进行一个验证。
代码全部都可以从GitHub - FADHLyemen/Metabolomics_signature 中找到。但其实真的在复现的时候,遇到了几个卡住的坑。
第一坑:配置环境
library(vegan)library(dplyr)library(cluster)library(pheatmap)library(siggenes)library(pamr)library(factoextra)library(ggplot2)library(MetaboAnalystR)library(tidyverse)library(ggpubr)
这一步应该很多人复现的时候就做不出来。其中有些包的安装会有问题。现在就以MetaboAnalystR为例,这个包无法通过install.packages,也无法通过BiocMananger::install这两种方式安装,需要从github上想办法去安装。
一种是通过devtools::install_github 安装,但目前大部分我的读者应该无法方便的通过这种方式安装。如果你可以使用一些服务,或者通过改变你所处地点等方式完成,拿着就不再是问题了。
另一种是找到github上MetaboAnalystR这个仓库,下载tar.gz那个文件(这个大陆可能会比较慢,但不是不能做到),通过yulab.utils::install_zip进行安装。如果提示缺少一些依赖包,先安装依赖包再安装。
然后就看到代码这里有写读一个文件,这文件怎么找呢?
一个是看到他代码里写道来源之一:
但这个下载下来看了下样子和他示范的样子不太一样。
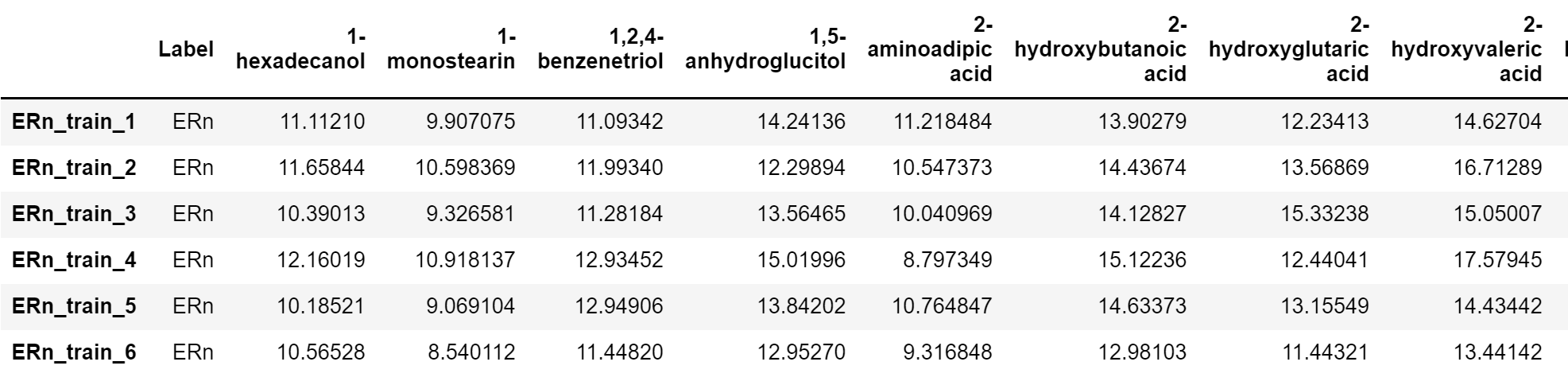
需要你自己处理一下(处理部分的代码他没有给出),这个自己探索如果没有基础可能会比较烦。我这里就抛砖引玉一下:
datread <- fread("./dataset1.xls")dat <- datread[,-2:-6]colname <- dat$metabolitedat <- dat[,-1]dat <- t(dat)colnames(dat) <- colnamedat <- as.data.frame(dat)dat$Label <- "No"dat[grep("^ERn_",rownames(dat)),]$Label <- "ERn"dat[grep("^ERp_",rownames(dat)),]$Label <- "ERp"dat <- dat[,c(163,1:162)]write.csv(dat,"./mmc1homemade.csv")
还好这github里,他还是把整理好的文件抛出来了。就下载下来直接弄也可以。
第二坑:代码包不配套
运行到后面,又发现问题了。这个函数ImputeVar我没有。搜索引擎一查这个函数是MetaboAnalystR这个包里的,但我安装了怎么还没有呢?我里面只有ImputeMissingVar这个函数。改一下名字一样也能用。但是再往下这一步运行一直报错:
mSet<-Normalization(mSet, "QuntileNorm", "LogNorm", "AutoNorm", ratio=FALSE, ratioNum=20)
这咋整?我回过头去看了下ImputeVar这个函数,是MetaboAnalystR这个第二版里面的函数,那对应的,文章给的代码自然就是通过第二版的流程进行。这里可以卸载了,直接安装对应版本的包(这里留个小尾巴,大家可以看看怎么做)。也可以去看下第三版的流程说明书:

里面找到了对应的流程,稍作修改:
mSet <- InitDataObjects("conc", "stat", FALSE)mSet <- Read.TextData(mSet, "mmc1.csv", "rowu", "disc")mSet <- SanityCheckData(mSet)mSet <- ReplaceMin(mSet)mSet <- FilterVariable(mSet, "none", "F", 25)mSet <- PreparePrenormData(mSet)mSet <-Normalization(mSet,"QuntileNorm","LogNorm","AutoNorm",ratio = FALSE,ratioNum = 20)mSet <- PlotNormSummary(mSet, "norm_0_", "png", 72, width = NA)mSet <- PlotSampleNormSummary(mSet, "snorm_0_", "png", 72, width = NA)Normalized_data = mSet$dataSet$normNormalized_data = Normalized_data[order(match(rownames(Normalized_data), rownames(prostate_df))), ]mydata = Normalized_data
这一关算是跨过去了。
最后一坑:验证数据集
对一个公共数据集GSE59198去下载处理的时候,也会遇到和安装包类似的困难。
这个坑避过是因为之前做过类似的工作,可以用yulab的工具做一个处理,使用GEOmirror里的geoChina来处理下载数据时遇到的问题。
改进后的代码块如下:
library(GEOmirror)gset <- geoChina("GSE59198")lumi.N <- lumiN(gset[[1]])e <- exprs(lumi.N)min(e)e1=log2(e-(min(e)+0.01))if (length(gset) > 1) idx <- grep("GPL16807", attr(gset, "names")) else idx <- 1gset1 <- gset[[idx]]colnames(e1)=gset1[[1]]ER=as.character(gset1[[11]])HER2=as.character(gset1[[12]])genes_expression_154=data.frame(t(e1),ER=ER,HER2=HER2)
到此为止,对这篇文章复现时候应该踩坑到此为止了(可能有其他的,只是我没想到和遇到,如果你们遇到了欢迎交流)
