@agpwhy
2021-11-23T03:50:30.000000Z
字数 1997
阅读 385
王胖的生信笔记第27期:序列比对
在正文开始前,先提点别的。前段时间搞了个视频整了个活儿(https://www.bilibili.com/video/BV1XS4y197xW),投了两个地方总点击也算过千了。以后考虑准备做成常规栏目,不过还是需要调整精进。对于视频制作,尤其是短视频制作我还没有太多的经验,希望大家多多批评指正。
故事的由来
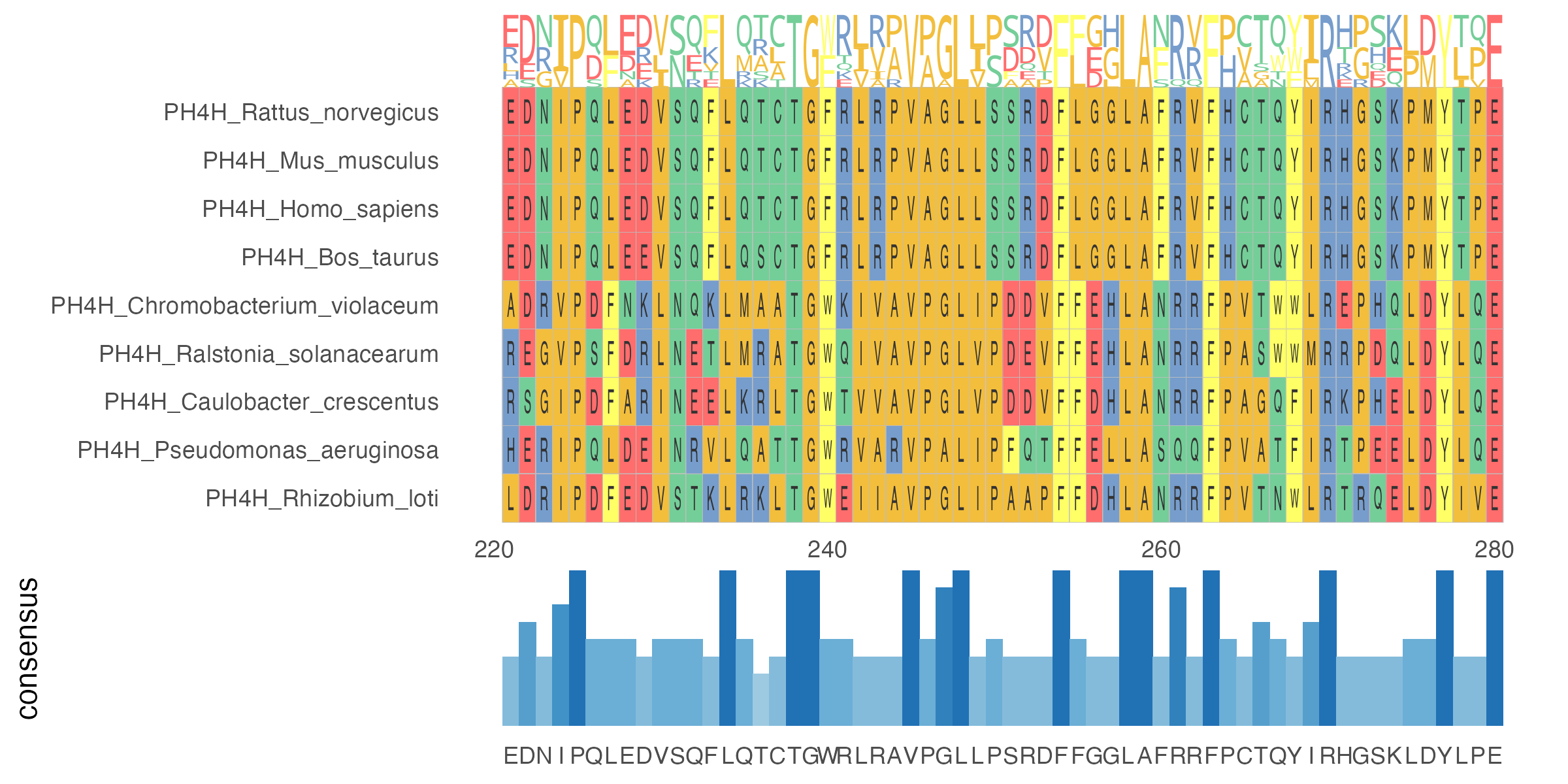
好了,回归主题,今天要写的一个笔记是一个很古早的包的运用(2015年的包)。为啥要讲这么古早的一个东西呢?因为这一周已经有两个人来问同一个事情了。如何做蛋白序列的比对呢?就类似这样的一个效果。【具体的做的什么东西的比对我就不写啦,涉及到来交流的人的个人问题】

其实最简单的方法就是上uniport,直接利用它们的align进行。但是这个出来的结果没这么好看。

怎么弄的更好看呢?其实有很多种fan个发,我这里就抛砖引玉,用一个2015年就有的工具包来进行一个展示。
准备
需要安装msa包。
BiocManager::install("msa")
同时准备好latex的texshade包
system.file("tex", "texshade.sty", package="msa")
我这里就用他的示范数据里。自己要弄比对蛋白序列的话,需要fasta格式的文件。一般来说,常见的都可以上uniport去下载。可以参考这篇https://blog.csdn.net/weixin_40408680/article/details/105607394。
mySequenceFile <- system.file("examples", "exampleAA.fasta", package="msa")
mySequences <- readAAStringSet(mySequenceFile)
myFirstAlignment <- msa(mySequences)
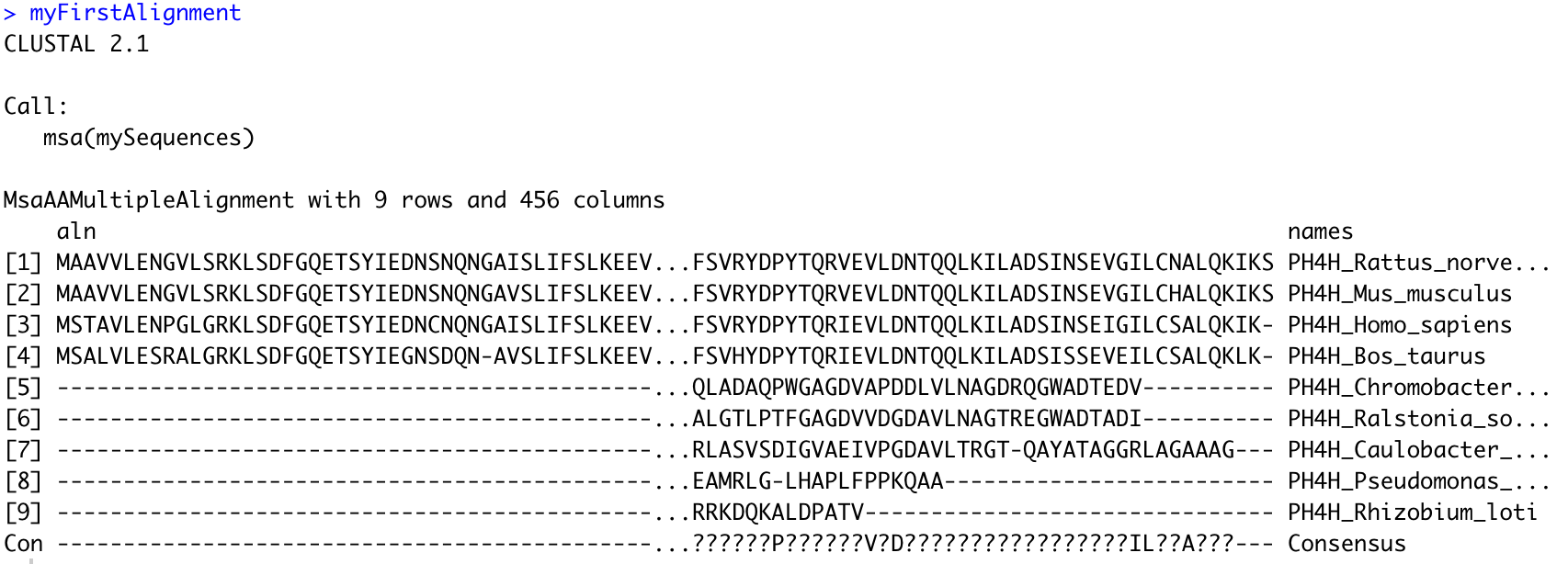
msaPrettyPrint(myFirstAlignment, output="pdf", showNames="none",showLogo="none", askForOverwrite=FALSE, verbose=FALSE)
然后写到这里突然发现,后面出问题了。
https://stackoverflow.com/questions/68195033/how-to-solve-this-error-when-run-latex-in-r
看了下可能和我LaTex编译器版本升级了有关。哎,那咋整呢?要么退环境要么换个方式。
展示翻车同时也得展示怎么解决翻车吧。
换条路
经过百度和同学指点,有个叫在msa包功能上有所进化的包ggmsa。
BiocManager::install("ggmsa")
library(ggmsa)
library(ggplot2)
就用包里自带的一些示例数据好了。
protein_sequences <- system.file("extdata", "sample.fasta", package = "ggmsa")
miRNA_sequences <- system.file("extdata", "seedSample.fa", package = "ggmsa")
nt_sequences <- system.file("extdata", "LeaderRepeat_All.fa", package = "ggmsa")
最简单的比对
ggmsa(protein_sequences, 300, 350, color = "Clustal", font = "DroidSansMono", char_width = 0.5, seq_name = TRUE )
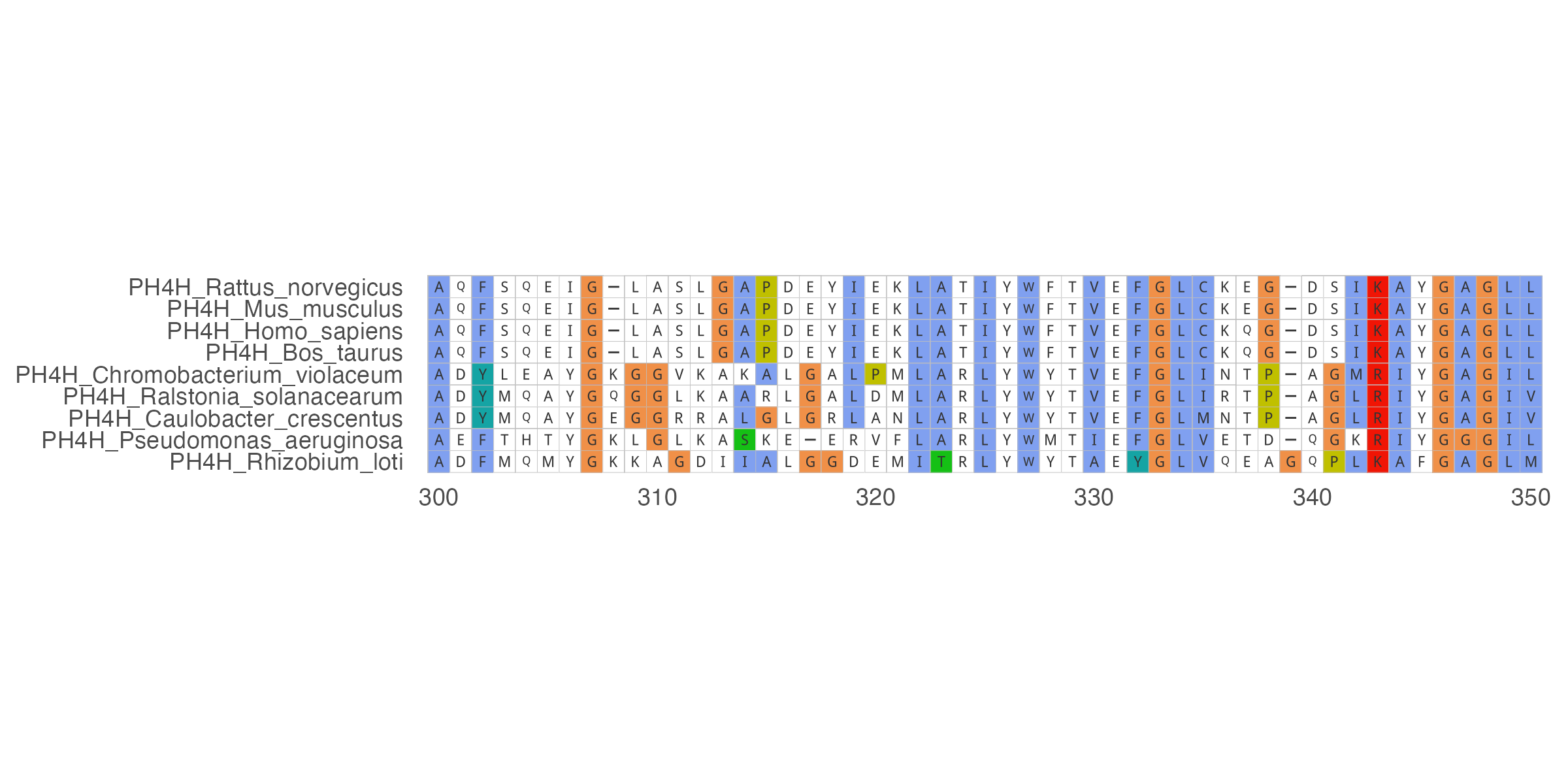
换个展示方式
ggmsa(nt_sequences,font = NULL, color = "Chemistry_NT") + geom_seqlogo(color = "Chemistry_NT") + geom_GC() + theme(legend.position = "none")
其他展示特定保守部分的方式。
ggmsa(miRNA_sequences, char_width = 0.5, color = "Chemistry_NT") + geom_seed(seed = "GAGGUAG", star = TRUE)

ggmsa(miRNA_sequences, char_width = 0.5, seq_name = TRUE, none_bg = TRUE) + geom_seed(seed = "GAGGUAG")

ggmsa(RF03120_msa, font = NULL, color = "Chemistry_NT", seq_name = F, show.legend = F, border = NA) +geom_helix(helix_data = RF_arc) + theme(axis.text.y = element_blank())
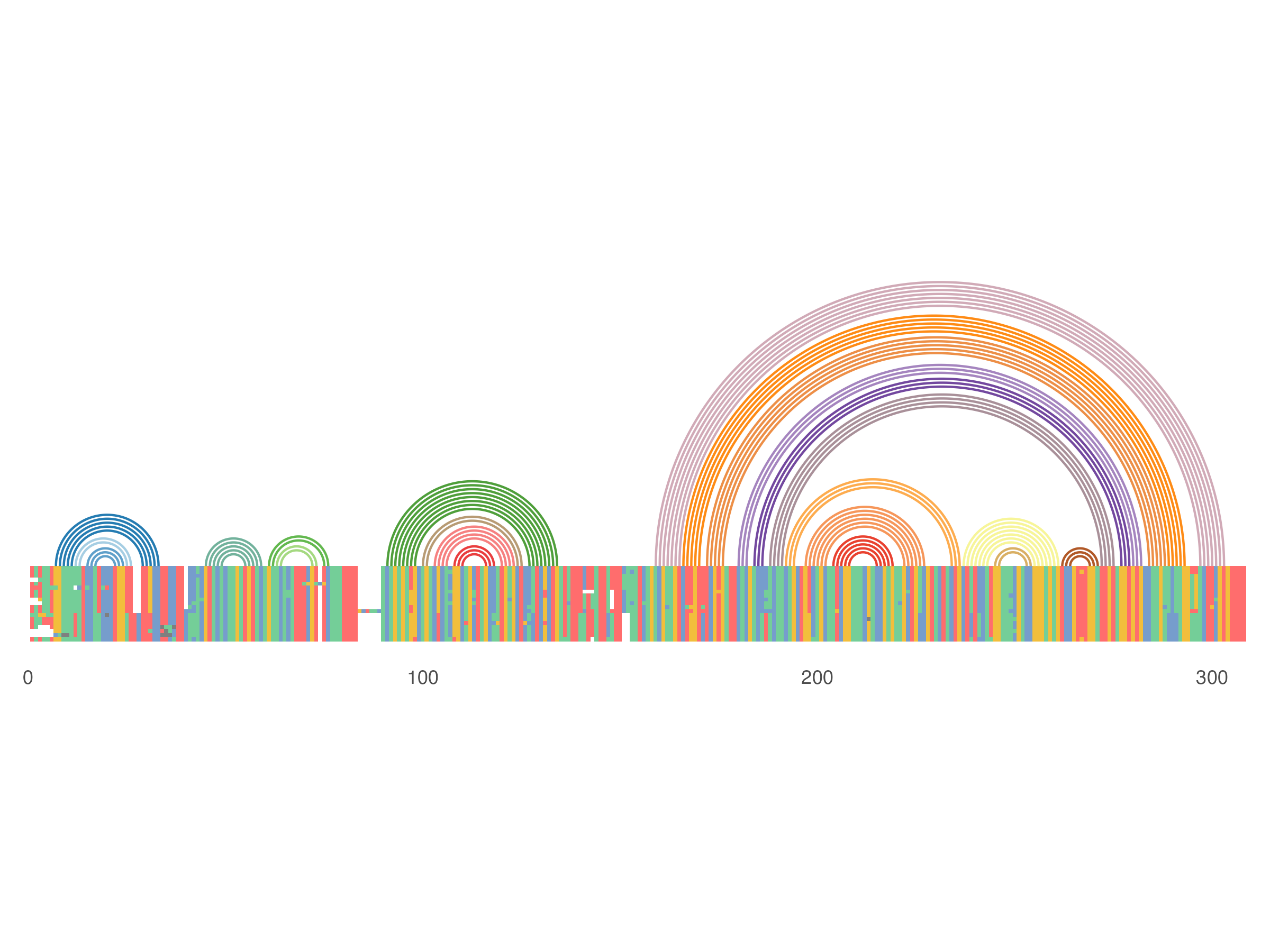
总之最后就可以有这样比较酷的msa展示方式了